Introduction
IntaRNA is a program for the fast and accurate prediction of interactions between two RNA molecules. It has been designed to predict mRNA target sites for given non-coding RNAs (ncRNAs) like eukaryotic microRNAs (miRNAs) or bacterial small RNAs (sRNAs), but it can also be used to predict other types of RNA-RNA interactions.
IntaRNA predicts RNA-RNA interactions by an energy-based approach that is based on two assumptions: (1) the accessibility of the interaction sites is important for the interaction formation, and (2) a seed region is required to initiate the interaction (e.g. the 5' seed region for miRNAs). In two previous studies on interactions between bacterial sRNAs and their target mRNAs, we presented evidence that the incorporation of these two requirements improves the prediction quality of IntaRNA (Busch et al., 2008; Richter et al., 2010).
The energy score of a predicted RNA-RNA interaction is the sum of the following contributions:
- the hybridization free energy of the interacting subsequences, and
- the free energies required to unfold the interaction sites in both RNA molecules.
Interactions are predicted by minimizing the interaction energy score via dynamic programming.
The free energy that is required to unfold the interaction site, i.e. making it accessible, is calculated from the thermodynamic ensemble of all secondary structures that can be formed by the RNA sequence. Ensemble free energy calculation is realized via the Vienna RNA library (Hofacker et al., 1994; Lorenz et al., 2011).
For the interaction between the two RNAs, IntaRNA requires the existence of an interaction seed. A seed is an initial interacting region of (nearly) perfect sequence complementarity, which is additionally often conserved. The user has to specify the minimal number of perfectly paired bases and the maximal number of unpaired bases in the seed region. Other seed features as the seed position in the ncRNA can be optionally defined by the user.
Precomputed results for
Enterobacteria:
ChiX,
CyaR,
FnrS,
GcvB,
MicC,
RyhB,
Spot42,
and for
Non-enteric bacteria:
LhrA2,
PrrF1,
Yfr1.
Note, in contrast to this server, the stand-alone IntaRNA software
does
not limit the problem size, provides enhanced functionality, and
offers a batch processing-friendly command line interface. For this reasons,
you might consider to
install IntaRNA locally
When using IntaRNA please cite :
- Martin Mann, Patrick R. Wright, and Rolf Backofen
IntaRNA 2.0: enhanced and customizable prediction of RNA–RNA interactions
Nucleic Acids Research, 2017, 45 (W1), W435–W439.
- Patrick R. Wright, Jens Georg, Martin Mann, Dragos A. Sorescu, Andreas S. Richter, Steffen Lott, Robert Kleinkauf, Wolfgang R. Hess, and Rolf Backofen
CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains
Nucleic Acids Research, 2014, 42 (W1), W119-W123.
- Anke Busch, Andreas S. Richter, and Rolf Backofen
IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions
Bioinformatics, 2008, 24 (24), 2849-56.
- Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.
Results are computed with IntaRNA version 3.4.1 linking Vienna RNA package 2.7.0
Overview
The following parameters are used to control the execution of IntaRNA
Furthermore, additional information is available
Sequence Parameters
 in FASTA") Query ncRNA (short) in FASTA
Query ncRNA (short) in FASTA
The query ncRNA is in general supposed to be the shorter of both
interacting RNAs. Note, no overlaps of reported interactions are
allowed for the target RNAs (but within the query RNAs).
IntaRNA accepts the input of query sequences in form of a multiple FASTA file. Input can be given either as direct text input or by uploading a file. Each sequence should contain only the characters A, C, G, T, U.
A sequence in FASTA format begins with a single-line sequence identifier that starts with a greater-than (">") symbol, followed by lines of sequence data. For readability, it is recommended that each line is at most 80 characters in length.
Ambiguous nucleotide(s) 'N' are excluded from all base pairings of the interaction.
The parameter constraints are: The input has to be in valid FASTA format. The number of sequences has to be at least 1 and at most 100. Sequence lengths have to be in the range 4-2000. The allowed sequence alphabet is 'ACGUTNacgutn'. In NCBI target mode, only 1 query sequence with length up to 750nt is allowed.
Defaults to ()
Index first query position
Index of first query sequence position. If not present, it defaults to 1. Setting to a negative number X will result in an indexing 'X,..,-1,+1,..(length+X)' omitting '0', as eg used for position information relative to an mRNA start codon.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to -9999999 and must be smaller than or equal to 9999999.
Defaults to (1)
Target RNA (long) in FASTA
The target RNA is in general supposed to be the longer of both
interacting RNAs. Note, no overlaps of reported interactions are
allowed for the target RNAs (but within the query RNAs).
IntaRNA accepts the input of target sequences in form of a multiple FASTA file. Input can be given either as direct text input or by uploading a file. Each sequence should contain only the characters A, C, G, T, U.
A sequence in FASTA format begins with a single-line sequence identifier that starts with a greater-than (">") symbol, followed by lines of sequence data. For readability, it is recommended that each line is at most 80 characters in length.
Ambiguous nucleotide(s) 'N' are excluded from all base pairings of the interaction.
The parameter constraints are: The input has to be in valid FASTA format. The number of sequences has to be at least 0 and at most 100. Sequence lengths have to be in the range 4-2000. The allowed sequence alphabet is 'ACGUTNacgutn'. The minimal sequence number is 1.
Defaults to ()
Target NCBI RefSeq ID
For
prokaryotes,
mRNA sequences can be automatically extracted from a genome instead of manual sequence input.
For a given NCBI RefSeq genome accession number, subsequences of each gene annotated in the genome are extracted. The accession number has to start with "NC_" followed by six digits or "NZ_" followed by some string, which refers to complete genomic molecules including genomes, chromosomes, and plasmids.
Each extracted subsequence consists of a user-specified number of nucleotides upstream and downstream of the start codon or the stop codon. Downstream positions start at start codon or after stop codon, respectively.
To check if the organisms you selected compatible,
check this
list of RefSeq IDs.
The list is regularly updated.
Please contact us if you know your organism is part of the RefSeq database
and has an ID in the NZ_* or NC_XXXXXX format but is not present in this list, or
is missing IDs. Then we can run an update.
The parameter constraints are: Access to the NCBI server is needed. Either this parameter or Target RNA (long) in FASTA can be set, not both at the same time. The given ID has to be in the list of supported RefSeq IDs (see help).
Defaults to ()
All replicons
Specifies whether or not IntaRNA should be executed on all replicons for the given NCBI RefSeq ID.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0 and must be smaller than or equal to 1.
Defaults to (1)
Extract sequences around
This option allows you to select from which region of the mRNAs
you would like to retrieve your putative target sequences. Selecting
"start codon" selects regions upstream and downstram (see nt up, nt down)
relative to the start codon.
The same logic holds if you select "stop codon".
nt upstream (1-300)
This parameter specifies the number of nucleotides (nt) upstream of your
start or stop codon (depending which one you selected). If you selected
start codon, and have prior knowledge about average 5'UTR lengths in your
input organisms then it is sensible to set nt up to this number in order
to increase prediction quality. The sum of nt up and nt down must be at
least equal to the window size.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 300.
Defaults to (75)
nt downstream (1-300)
This parameter specifies the number of nucleotides (nt) downstream of your
start or stop codon (depending which one you selected). If you selected
start codon, and have prior knowledge about average 3'UTR lengths in your
input organisms then it is sensible to set nt up to this number in order
to increase prediction quality. The sum of nt up and nt down must be at
least equal to the window size.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 300.
Defaults to (75)
Index first target position
Index of first target sequence position. If not present, it defaults to 1.
Setting to a negative number X will result in an indexing 'X,..,-1,+1,..(length+X)' omitting '0',
as eg used for position information relative to an mRNA start codon.
Thus, in NCBI mode, you can set it to the negated '-NtUpstream' to get a start-codon-based indexing of the target RNAs.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to -9999999 and must be smaller than or equal to 9999999.
Defaults to (1)
Output Parameters
Number of interactions per RNA pair
Maximal number of (sub)optimal interactions that are predicted per RNA pair. Select overlapping constraints for suboptimal interactions.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 100. In NCBI mode, this parameter has to be '1.0'.
Defaults to (5)
Suboptimal interaction overlap
Defines where overlapping interactions in the suboptimal output are allowed: N - no overlaps at all, T - overlaps in target only, Q - overlaps in query only, B - overlaps in both query and target.
Max. interaction length
Output only interactions that involve subsequences of
lengths up to the given threshold. Set 0 or leave free to disable the constraint.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0.
Defaults to ()
Max. absolute energy of an interaction
Output only interactions with an energy below or equal to this energy in kcal/mol.
Note, if the minimum free energy (mfe) of any interaction is above
this threshold, no interaction will be reported.
The parameter constraints are: Input value has to be parsable as Double. The value must be smaller than or equal to 0. In NCBI mode, this parameter has to be '0.0'.
Defaults to (0)
Max. delta energy above mfe of an interaction
Output only interactions with an energy below or equal to the minimum free energy (mfe) + this delta energy term in kcal/mol.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 100.
Defaults to (100)
No lonely base pairs
If enabled, no unstacked (lonely) base pairs are considered for prediction.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
No GU at helix ends
If enabled, no GU base pair is allowed at helix ends (ie. interaction ends or within loops).
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
Seed Parameters
Min. number of basepairs in seed
Minimal number of intermolecular base pairs in the seed region. Note, for webserver use this value is restricted.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 2 and must be smaller than or equal to 12. Has to be greater than the number of unpaired bases within the seed (for webserver use). Has to be as long as the minimal seed length (seed base pairs).
Defaults to (7)
Max. Number of mismatches in seed
Maximal number of unpaired bases in the seed region in both sequences. Note, for webserver use this value is restricted.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0 and must be smaller than or equal to 20.
Defaults to (0)
Maximal energy
Maximal overall interaction energy of the seed interaction to be considered for further interaction prediction. Note, a value too low will discard all seeds and thus result in no predicted interaction. A value too high will cause weaken the seed constraint.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to -999 and must be smaller than or equal to 999.
Defaults to (0)
Minimal unpaired probability (per RNA)
The minimal unpaired probabilitiy of the seed interaction sites (checked independently for query and target seed site)
to be considered for further interaction prediction. Note, a value too high will discard all seeds and thus result in no predicted interaction.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 1.
Defaults to (0)
Seed position (query)
Seed search and position is constrained to this region of the query ncRNA. The start and the end position of the region have to be given in 5' to 3' direction of the RNA starting from position 1.
The parameter constraints are: Has to be in the format 'FROM-TO' to give the coordinates FROM where TO where the seed is to be found (index counting starts with 1).
Defaults to ()
Seed position (target)
Seed search and position is constrained to this region of the target RNA. The start and the end position of the region have to be given in 5' to 3' direction of the RNA starting from position 1.
The parameter constraints are: Has to be in the format 'FROM-TO' to give the coordinates FROM where TO where the seed is to be found (index counting starts with 1).
Defaults to ()
Ignore seeds with GU base pairs
If enabled, no seeds containing GU base pairs are considered for prediction.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Ignore seeds with GU ends
If enabled, no seeds with GU base pairs at their ends are considered for prediction.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
Folding Parameters
Temperature for energy computation
Temperature in degrees Celsius used to rescale energy parameters.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 0.
Defaults to (37.0)
Access. query: folding window size
Size of the averaging window in the local query
ncRNA folding (RNAplfold -W) for the computation of accessibilities.
Local folding is key to reasonable folding results when facing
larger RNA molecules, since it minimizes effects of incorrect
long-range predictions (see
local folding article).
Note, the folding window size should be about 50nt higher than the
max. basepair distance.
If set to 0, no sliding window is used and the full sequence length is considered.
The same holds, if the value is larger than the sequence length.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0. The window size has to be at least as large as the maximal basepair distance.
Defaults to (150)
Access. query: max. basepair distance
Maximal distance of two paired bases in the local
query ncRNA folding (RNAplfold -L) for computation of accessibilities.
Local folding is key to reasonable folding results when facing
larger RNA molecules, since it minimizes effects of incorrect
long-range predictions (see
local folding article).
Note, max. basepair distance should be about 50nt less than the
the folding window size.
If set to 0, the sliding window size value is also used for base pair span restrictions.
The same holds, if the value is larger than the sequence length.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0.
Defaults to (100)
Access. target: folding window size
Size of the averaging window in the local target
RNA folding (RNAplfold -W) for the computation of accessibilities.
Local folding is key to reasonable folding results when facing
larger RNA molecules, since it minimizes effects of incorrect
long-range predictions (see
local folding article).
Note, the folding window size should be about 50nt higher than the
max. basepair distance.
If set to 0, no sliding window is used and the full sequence length is considered.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0. The window size has to be at least as large as the maximal basepair distance.
Defaults to (150)
Access. target: max. basepair distance
Maximal distance of two paired bases in the local
target RNA folding (RNAplfold -L) for computation of accessibilities.
Local folding is key to reasonable folding results when facing
larger RNA molecules, since it minimizes effects of incorrect
long-range predictions (see
local folding article).
Note, max. basepair distance should be about 50nt less than the
the folding window size.
If set to 0, the sliding window size value is also used for base pair span restrictions.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0.
Defaults to (100)
Energy parameter set (Vienna package)
Defines what energy parameter set to be used within
the Vienna RNA package to compute base pair probabilities,
i.e. dot plots, for each input sequence. The parameter sets are
provided by the Vienna RNA package (see version information) and are
named according to the first author and year of the corresponding
publication.
Output Description
The output table summarizes the
best 100 predicted interactions. It can be sorted by clicking on the header of a column.
NOTE: If your sequence input combinatorics exceeds 100, the output will not cover all possible combinations!
For the selected table row, the according interaction is shown below the table along with additional information such as interaction positions, different contributions of the interaction energy score, etc.
Details on the information:
- Energy : Energy score of the interaction in kcal/mol = sum of the energy of the hybridisation and the two unfolding energy terms. They are subjected to length normalization, when running in NCBI mode with coding sequence regions.
- Position : Positions are given in 5' to 3' direction starting from position 1.
- Seed Position : Location of the most stable putative seed subinteraction. Note, an interaction can feature multiple putative seeds! The webserver reports only the seed with lowest overall energy (if only seed is formed).
- Unfolding Energy : Free energy in kcal/mol that is required to make the interaction site accessible. Global folding of the query RNA and local folding of the target RNA is assumed.
- Hybridisation Energy : Hybridization free energy in kcal/mol of the interacting RNA subsequences.
Furthermore in NCBI mode:
- Target sequences : The target sequences downloaded for the given NCBI RefSeq ID are accessible vi the input parameter link for 'Target RNA in FASTA'.
- Functional enrichment file : Functional enrichment for the top 50 predictions employing DAVID annotation.
- p-value : Statistical probability to calculate an interaction with an energy smaller or equal to the found one.
- fdr value : p-value corrected for multiple testing (see FAQ).
- Region plots : Visualization of important interaction regions (see below).
Functional Annotation Chart:
The top 50 targets have been subjected to functional
enrichment. The heatmap shows all members of clusters with a
DAVID
enrichment score >= 1 in a specific color. Each row represents a gene and
each column a specific functional term. If the gene can be assigned to a
term, the corresponding square is filled/colored. Closely related terms are
assigned to a cluster and have the same color. The opacity of the
color depends on the p-value of the IntaRNA prediction. A more intense
color represents a more significant p-value. The "Fold enrichment" is
given in front of the term descriptions. It gives the enrichment of a
term in the prediction group in relation to the whole genome background
(e.g. a term with an enrichment of 10 contains 10 times more genes
belonging to the respective term than the background). The enrichment
scores give a measure of the biological significance of the cluster. A
higher score represents a more statistically significant enrichment. The
publication on the
DAVID
webserver suggests to investigate clusters with an
enrichment score of >= 1.3.
Downloadable files in NCBI mode:
Functional Enrichment:
This file contains the
DAVID
functional enrichment result
for the target candidates up to IntaRNA p-values <= 0.01.
A certain term appears as enriched, if it is significantly
overrepresented in the top list when compared to the background.
The background in this case are all genes for which there is
a prediction (not the entire set of genes of an organism).
Enrichment scores of 1.3 and higher, suggest
statistical significance. However, enrichments also strongly
depend on the quality of the annotation of the entered organism of interest.
The file is tab delimited. This result is only calculated for
the organism of interest.
Regions plots:
These plots are meant to give you an overview of the regions in the
target and query sequences that play predominant roles.
It is based on the top-25 predicted targets.
Since targets are extracted from the genomic context around start
or stop codons, the respective position is highlighted by a vertical
line.
Input Examples
pairwise predictions
pairwise ncRNA-mRNA predictions
The
example's result can be directly accessed
here PrrF1 - Pseudomonas aeruginosa
PrrF1 - Pseudomonas aeruginosa
The
example's result can be directly accessed
hereFrequently Asked Questions
If your question is not listed, please send it to us!
 My sequences are longer than allowed within the webserver input, what can I do?
My sequences are longer than allowed within the webserver input, what can I do?
The restriction of the RNA input length (and number) is due to the limited resources
available for the webserver computations. In order to deal with large(er)
inputs your can
(A) install
IntaRNA locally (recommended)
(B) chunk up your long sequences into overlapping subsequences, e.g. using
|---------------------------| = full sequence
|--------------|------------| = first chunk set
|-------|-------------|-----| = second chunk set
Given the (two) sets: (1) run independent predictions, (2) merge the output lists,
(3) check the top ranking results.
You might have to manually remove/ignore high-ranking hits close to the sequence's cut points (less than 150nt),
since the accessibility computation of these regions is strongly biased
by the sequence decomposition. See below for a graphical depiction of an alternative strategy.
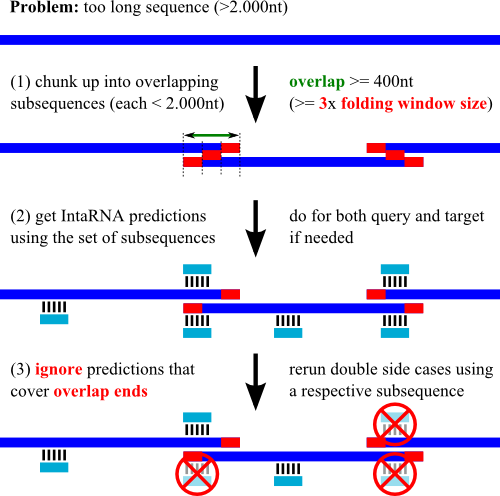
How does the mRNA length influence the energy score reported by IntaRNA?
IntaRNA is based on minimization of an energy score that incorporates the hybridization energy and the accessibility of the interaction sites in both RNAs. The hybridization energy alone depends only the length of the interaction. For computation of accessibilities in the mRNA, it is assumed the mRNAs are folded locally allowing only base pairs with the given maximal span. The accessibilitiy for the interaction site in the mRNA is averaged over all windows of the given size that contain the interaction site.
How is accessibility defined?
The accessibility of the interaction site is the free energy that is required to make it single stranded. It is defined as the difference between the free energy of the ensemble of all RNA secondary structures and the free energy of the ensemble of RNA secondary structures, where the interaction site is single stranded.
How are accessibilities calculated?
The calculation of accessibilities is based on ensemble free energies. Ensemble free energies are calculated using a partition function approach (McCaskill, 1990) assuming global folding of the ncRNA and local folding of the mRNA. For this purpose, RNAplfold and RNAup are integrated into IntaRNA via the Vienna RNA library (Hofacker et al., 1994; Bernhart et al., 2006, Mückstein et al., 2008).
What are the fdr values and how to interpret them?
The fdr (false discovery rate) values are most easily explained with an example. Assume a fdr
cutoff of 0.5. Statistically speaking, 50% of all predicted targets in the
list up to this cutoff are assumed to be false positives. The fdr
gives you an impression of how many incorrect predictions to expect up to
a certain threshold. The fdr values are computed using the R-function
p.adjust
and the method by (Benjamini&Hochberg, 1995).
Where can I see/download the used target RNAs derived for my NCBI RefSeq ID?
The target sequences downloaded from NCBI for the given RefSeq ID
are available for download in FASTA format in the 'Input Parameter'
section of the result page. The FASTA file is linked for parameter
'Target RNA in FASTA'.
The putative targets are sorted in the reverse order in the regions plot
when compared to the main result table. Which sorting should I trust?
The reverse sorting in the regions plots is due to our plotting script.
This means that you should trust the initial sorting of the main result
table.
List of Changes
- 5.0.11 - 250208 : IntaRNA v3.4.1 : version update
- 4.8.0 - 200302 : IntaRNA v3.2.0 : bugfix of accessibility constraint usage
- 4.7.2 - 200131 : IntaRNA v3.1.5 : bugfix of predictions using seeds with bulges and noLP constraint
- 4.7.1 - 200131 : IntaRNA v3.1.4 : bugfix of predictions using seeds with bulges
- 4.7.0 - 200128 : IntaRNA v3.1.3 : version updated (faster, rounded E values, more robust) and backend refactored; new parameters; !!! DEFAULTS CHANGED !!! (outNoLP, outNoGUend, seedNoGUend)
- 4.5.8 - 180906 : IntaRNA v2.3.0 : faster seed screening
- 4.5.7 - 180904 : IntaRNA v2.2.1 : uses now Vienna RNA package v2.4.9. Bugfixes in the Vienna RNA package cause changes in the window-based accessibility computation and thus may yield different results than before
- 4.5.5 : Bugfix: number of suboptimal interactions was ignored (introduced in v4.5.2)
- 4.5.1 : IntaRNA v2.1.0 : uses now Vienna RNA package v2.4.3. Bugfixes in the Vienna RNA package cause changes in the window-based accessibility computation and thus may yield different results than before
- 4.4.8 : IntaRNA v2.0.5 (bugfix release)
- 4.4.3 : IntaRNA v2.0.2 (bugfix release)
- 4.4.2 : IntaRNA v2.0.1 (local query accessibility computation (W=150,L=100); mininmal energy profile / heatmap enabled) (wrapper v1.1.2)
- 4.4.1 : interface enables setup where (suboptimal) interactions are allowed to overlap
- 4.4.0 : IntaRNA v2.* (wrapper v1.1.1) online
- 4.0.9 : 'coding sequence' region option removed since resulting sequences are usually too long to be supported by the web server backend. Please contact us if you are interested how to extract such sequences yourself.
- 4.0.8 : IntaRNA wrapper v1.0.7.1 online : changed DAVID-WS from perl to python client
- 4.0.0 : IntaRNA wrapper v1.0.7 online : Added help and exception handling to wrapper backend
- 3.5.0 : IntaRNA wrapper v1.0.6 online : using local mirrors of old NCBI ID system for compatibility if available
- 3.4.1 : IntaRNA wrapper v1.0.5 online : support of new NCBI ID system
- 3.3.7 : Maximal number of input sequences is now 100 x 100. Note: only the best 100 interaction predictions reported!
- 3.3.5 : IntaRNA wrapper v1.0.4 online : Now using (Benjamini&Hochberg, 1995) for false discovery rate (fdr) estimation
- 3.3.0 : FASTA input is allowed to contain unspecified 'N' nucleotide characters
- 3.1.0 : for NCBI mode: p/q-values, functional enrichment
