
- Martin Mann, Mostafa M Mohamed, Syed M Ali, and Rolf Backofen
Interactive implementations of thermodynamics-based RNA structure and RNA-RNA interaction prediction approaches for example-driven teaching
PLOS Computational Biology, 14 (8), e1006341, 2018. - Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.
Teaching RNA algorithms
In order to enable an example-driven learning and teaching of
RNA structure related algorithms, we provide here Javascript-based
implementations for various algorithms for RNA structure and
RNA-RNA interaction prediction. To reduce the level of complexity,
all algorithms use a simple Nussinov-like energy scoring scheme, i.e.
the energy of an RNA structure is directly related to its number
of base pairs without further distinction.
Furthermore, we provide interactive implementations for general sequence alignment algorithms that are taught in our bioinformatics courses.
The source code of all implementations is available @github.com/BackofenLab/RNA-Playground.
Furthermore, we provide interactive implementations for general sequence alignment algorithms that are taught in our bioinformatics courses.
The source code of all implementations is available @github.com/BackofenLab/RNA-Playground.

RNA structure
Using an adapted umambigous version of the Nussinov algorithm
it is possible to count the number of all possible nested
secondary structures an RNA molecule can form.
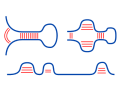
The Nussinov algorithm enables the efficient computation of
the structure with the maximal number of base pairs for a given
RNA sequence.
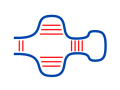
The McCaskill algorithm enables the efficient computation
of RNA structure probabilities as well as probabilities
that a certain base pair is formed. Furthermore, unpaired
probabilities for subsequences can be computed that
reflect the accessibility of RNA parts for other interactions.

The maximum expected accuracy (MEA) algorithm uses base pair
probabilities as well as unpaired probabilites (e.g. computed
via the McCaskill algorithm) to find the structure that is
'maximally accurate' in its structural elements.
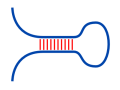
RNA-RNA interaction
RNA-RNA interactions can be predicted by linking the two
interacting RNAs into one pseudo-RNA and use a variant of
the Nussinov algorithm to predict the fused structure.
Such approaches are called 'co-folding'.
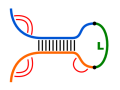
Given two RNA molecules, the RNA-RNA interaction with the
maximal number of intermolecular base pairs can be predicted.
Such algorithms ignore the competition between intra- and
intermolecular base pair formation, e.g. considered in
'co-folding' or 'accessibilty-based' approaches.
Since a nucleotide can form at most one base pair, there is
a competition between intra- and intermolecular base pair
formation. This can be incorporated into RNA-RNA interaction
prediction by also scoring the probability that an interacting
subsequence is not involved in intramolecular base pairs, i.e.
it is accessible.
Sequence alignment
S. B. Needleman and C. D. Wunsch introduced 1970 the first dynamic programming
based algorithm for the efficient global alignment of two
sequences. The approach uses a linear gap scoring scheme.

The global pairwise alignment scheme by Needleman and Wunsch
requires quadratic memory to store the dynamic programming
data structures. D. S. Hirschberg introduced 1975 an adaption
of the computation strategy in order to reduce the memory
requirement to a linear value while keeping the original runtime.

M. S. Waterman, T. F. Smith and W. A. Beyer introduced 1976
a variant of the global Needleman-Wunsch alignment approach that allows for
arbitrary gap scoring. While more general, the approach
has a higher computational complexity compared to the
Needleman-Wunsch method.

O. Gotoh introduced 1982 an efficient global alignment
approach that enables a more realistic affine gap cost
model without changing the computational complexity compared
to the Needleman-Wunsch approach.

T. F. Smith and M. S. Waterman adapted 1981
the Needleman-Wunsch approach to local alignment with linear
gap scoring. This enables the identification of most
similar subsequences.

O. Gotoh introduced 1982 an efficient global alignment
approach that enables a more realistic affine gap cost
model without changing the computational complexity compared
to the Needleman-Wunsch approach. Here, we extend the
original approach to local alignments by applying the
ideas of the Smith-Waterman algorithm.

A. N. Arslan, Ö. Eğecioğlu and P. A. Pevzner
introduced 2001 a fractional-programming-based approach to
compute normalized local alignments.
That is, it identifies the local alignment that is best
preserved with respect to the length of the aligned subsequences.
This counters the problem of the Smith-Waterman
approach where often longer alignments hide shorter alignments
with less gaps since they enable higher scores.

The progressive multi-sequence-alignment (MSA) scheme introduced 1987 by
D.-F. Feng and R. F. Doolittle is a heuristic approach to
compute close-to-optimal MSAs in reasonable time. This is
achieved by progressively joining subsets of sequences to
MSAs based on pairwise alignments, which is following by a
distance-based clustering tree. To this end, similarity
scores of pairwise alignments are converted into distance
scores needed for the clustering.

Progressive multi-sequence-alignment (MSA) schemes are fast but
only heuristics and thus are not always providing the optimal
MSA. Their result quality can be improved when iterative
refinement techniques are applied.

Progressive alignment schemes like the Feng-Doolittle approach
can show suboptimal alignment steps since they can be misguided
by the pairwise alignments that define how the progressive
alignment fusions are performed. This is tackled by the
t-coffee approach introduced 2000 by
C. Notredame, D. G. Higgins and J. Heringa. Instead of using
single pairwise alignments, the t-coffee approach derives
an input-specific scoring using an arbitrarily large set of
input alignments. This scoring scheme is sequence-position-specific
and thus encodes what sequence positions are (most often)
aligned in the input and thus indirectly also encodes unaligned
positions, i.e. gaps. Furthermore, 'indirect' position alignment
identified by combinations of alignment triples are incorporated
and provide a consistency-focusing scoring extension. The
final scoring is used in a adapted progressive alignment
scheme to compute a final multiple sequence alignment.

Evol. Clustering
Different methods can be applied to derive an evolutionary
/ clustering tree from given pairwise distance data.

When using our resources please cite :
About this web server
|
version 5.0.12
|
Copyright © 2012 - 2025 Bioinformatics Group Freiburg
|
 |
Imprint and Disclaimer
|
Imprint and Disclaimer