Frequently Asked Questions
If your question is not listed, please send it to us!
Troubleshooting
 How long are computed results available and stored?
How long are computed results available and stored?
All jobs computed on the Freiburg Tools webserver are stored for 30 days. Afterwards they are automatically removed. In order to preserve your job results you might want to use the job zip-file that is offered for each job on the according result page. This file contains all input information, call details and output files generated.
What publications to cite when using the server?
Each tool offered by this web server comes with a specific list of publications. Please cite them when using the web server.
Why do I get a warning saying that my browser has incompatibility issues with the web server?
The Freiburg Tools web server is known to have serious compatibility issues with Internet Explorer. If you are using this browser to access it, we highly recommend switching to Mozilla Firefox so that you don't experience lack of functionality.
The web server was also tested under Google Chrome, Opera, Safari and Konqueror and they are all known to work fine.
If you are not using Internet Explorer and you still get the warning message, then browser mimicry could be the reason. Please let us know and we will correct the problem as soon as possible.
Who to contact if I have further trouble or encounter problems not listed here?
Please
contact us as soon as you encounter any problems or difficulties. If you have problems with a specific tool please send as much detail as possible. In case your problems are related to a certain jobs, please provide the job ID etc. Thanks for your help and feedback!
I have created a cool new RNA Bioinformatics tool, is it possible to integrate it into the web server?
The Freiburg Tools web server is a very flexible and generic platform to integrate new tools. So please
contact us and we will happily discuss the possibilities of an integration of YOUR TOOL into our web server.
General questions
How are RNA structures modelled?
RNA structures are modelled as secondary structures, ie. each nucleotide/base can participate in at most one base pairing.
Only Watson-Crick (G-C,A-U) and wobble (G-U) base pairs are considered.
A structure is typically represented by dot-bracket-string encodings, where an opening bracket denotes a
pairing with a nucleotide successive in the sequences and a closing bracket the contrary.
For crossing, pseudoknot structures, different bracket symbols are needed.
Why are mRNAs assumed to fold locally and not globally?
mRNAs are usually loaded with translating ribosomes, which are able to disrupt
structures in the mRNA coding sequence due to their helicase activity (Takyar et
al., 2005). Furthermore, long range base pairs in large RNA molecules are
kinetically unfavored (Flamm et al., 2000), and long range base pairs predicted
by energy minimization are very inaccurate (Doshi et al., 2004).
IntaRNA
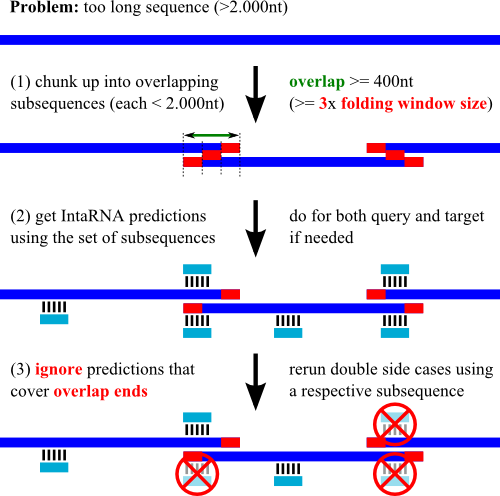
My sequences are longer than allowed within the webserver input, what can I do?
The restriction of the RNA input length (and number) is due to the limited resources
available for the webserver computations. In order to deal with large(er)
inputs your can
(A) install
IntaRNA locally (recommended)
(B) chunk up your long sequences into overlapping subsequences, e.g. using
|---------------------------| = full sequence
|--------------|------------| = first chunk set
|-------|-------------|-----| = second chunk set
Given the (two) sets: (1) run independent predictions, (2) merge the output lists,
(3) check the top ranking results.
You might have to manually remove/ignore high-ranking hits close to the sequence's cut points (less than 150nt),
since the accessibility computation of these regions is strongly biased
by the sequence decomposition. See below for a graphical depiction of an alternative strategy.
How does the mRNA length influence the energy score reported by IntaRNA?
IntaRNA is based on minimization of an energy score that incorporates the hybridization energy and the accessibility of the interaction sites in both RNAs. The hybridization energy alone depends only the length of the interaction. For computation of accessibilities in the mRNA, it is assumed the mRNAs are folded locally allowing only base pairs with the given maximal span. The accessibilitiy for the interaction site in the mRNA is averaged over all windows of the given size that contain the interaction site.
How is accessibility defined?
The accessibility of the interaction site is the free energy that is required to make it single stranded. It is defined as the difference between the free energy of the ensemble of all RNA secondary structures and the free energy of the ensemble of RNA secondary structures, where the interaction site is single stranded.
How are accessibilities calculated?
The calculation of accessibilities is based on ensemble free energies. Ensemble free energies are calculated using a partition function approach (McCaskill, 1990) assuming global folding of the ncRNA and local folding of the mRNA. For this purpose, RNAplfold and RNAup are integrated into IntaRNA via the Vienna RNA library (Hofacker et al., 1994; Bernhart et al., 2006, Mückstein et al., 2008).
What are the fdr values and how to interpret them?
The fdr (false discovery rate) values are most easily explained with an example. Assume a fdr
cutoff of 0.5. Statistically speaking, 50% of all predicted targets in the
list up to this cutoff are assumed to be false positives. The fdr
gives you an impression of how many incorrect predictions to expect up to
a certain threshold. The fdr values are computed using the R-function
p.adjust
and the method by (Benjamini&Hochberg, 1995).
Where can I see/download the used target RNAs derived for my NCBI RefSeq ID?
The target sequences downloaded from NCBI for the given RefSeq ID
are available for download in FASTA format in the 'Input Parameter'
section of the result page. The FASTA file is linked for parameter
'Target RNA in FASTA'.
The putative targets are sorted in the reverse order in the regions plot
when compared to the main result table. Which sorting should I trust?
The reverse sorting in the regions plots is due to our plotting script.
This means that you should trust the initial sorting of the main result
table.
LocARNA
ClustalW (or my favourite sequence alignment tool) is faster, why should I use LocARNA for aligning RNAs?
Sequence alignment programs like ClustalW, T-Coffee, MUSCLE, or MAFFT compare RNAs only by sequence similarity. However, for many RNAs the structure information is strongly conserved. Thus, sequence similarity can be weak even for closely related RNAs of the same family. Consequently, only sequence-structure alignment programs that consider sequence and structure similarity, such as LocARNA, can reveal their true similarity. Pure sequence alignment programs will usually completely fail if sequence identity drops below 60%. Unfortunately, fully taking structure information into account is computationally expensive. Nevertheless, LocARNA achieves very good performance for this class of algorithms.
Why should I use LocARNA compared to the RNA alignment program XY?
LocARNA is one of the fastest programs that do true sequence-structure alignment of RNAs and therefore produce highly accurate alignments. The LocARNA server is nice and offers a rich interface. You can even specify anchor constraints and structure constraints for global and local alignment; also probabilistic multiple alignment with alignment reliabilities is unique to LocARNA. Honestly, there are other very good programs out there; try them and compare!
How does LocARNA achieve its speed and low space consumption? Does this compromise accuracy?
LocARNA uses different heuristics for improving speed and lowering space requirements. Most importantly, it filters base pairs by their probability in the RNA structure ensemble and considers only 'significant' base pairs that pass a certain probability threshold. This reduces complexity from O(n^6) time and O(n^4) space to O(n^4) time and O(n^2) space, respectively. Furthermore, one can control which base pairs are compared at all by setting a maximal length difference. Finally, as reasonable for global alignment, one can control which residues are compared by LocARNA by a maximal difference of sequence positions. All those heuristics are optional and can be controlled in the advanced section of the server. However, the heuristics with default settings were shown to preserve alignment accuracy on the comprehensive Bralibase 2.1 benchmark. It's therefore likely that default settings won't compromise the alignment accuracy for your RNAs.
I am using LocARNA in local alignment mode; why does LocARNA return an empty multiple alignment (or only a very small one)?
Generally, LocARNA constructs multiple alignments by progressively merging (previously computed) alignments of fewer sequences. In the case of local alignment, LocARNA merges local subalignments from previous progressive alignment steps. In this way, the alignments can become shorter and shorter with every merge, since each progressive step retains only the sufficiently similar parts of the subsequences. Thus, if LocARNA cannot not find sufficiently similar common subsequences in all of your input sequences, it will produce a very small or even empty alignment.
Even in the latter case, LocARNA still computes the relations between all of your sequences in the form of a guide tree; furthermore it computes the alignments of the sequence subsets corresponding to the inner nodes of this tree. In many cases, one is rather interested in those alignments than in one single alignment of all sequences.
I want to align more sequences than allowed. What can I do?
Often it is a good idea to first look for high sequence similarity in
such a set of sequences. If one can identify sequence pairs with e.g.
more than 80% or 90% identity, usually it does not pay off to align them based
on structure similarity. Please check this by running a multiple
sequence alignment first.
If this is the case with your set of sequences, you could reasonably
reduce the number of sequences that you feed to our web server by
omitting extremely similar sequences.
I want to align longer sequences than allowed. What can I do?
An idea that might work: split the long sequence into (e.g. three)
overlapping "windows", i.e. subsequences, and use the web server.
This is especially useful, if your are aligning short sequences with
one (or few) long sequence(s) looking for a conserved motif.
RNAalifold consensus structure output shows single-letter codes, what is the meaning?
The consensus structure output is produced with RNAalifold's option '--mis', i.e. RNAalifold
outputs the "most informative sequence" instead of simple consensus: For each
column of the alignment output the set of nucleotides with frequence
greater than average in IUPAC notation
(see
RNAalifold manual page).
This means that the special letters are
IUPAC codes for nucleotides.
This is especially useful, if your are aligning short sequences with
one (or few) long sequence(s) looking for a conserved motif.
ExpaRNA
Can ExpaRNA compare multiple RNAs?
Currently, only pairwise comparison is supported. Maybe in future a multiple comparison is possible.
What is the difference between the scoring 'default' and 'prefer larger substructures'?
With the default scheme (t=1) ExpaRNA tries to find a set of common substructures with maximal number of nucleotides. With the alternative scheme (t=2), the score of each substructure is quadratic to its size in nucleotides. For example, a base pair (2 nucleoides) has score 4, two base pairs have score 16 and so on. This prefers larger substructures in the result.
Can ExpaRNA find substructures with mismatches?
No, ExpaRNA only finds exact matching substructures. Both sequence and structure of all nucleotides have to be exactly the same. If you are interested in a pure structural comparison you can change all nucleotides to 'N' and use the structure of the original sequence.
Can ExpaRNA do a structural comparison only?
Yes, to some extend. As a trick you can change all nucleotides to 'N' in combination with the structure of the original sequence.
CRISPRmap
Where can I download the orginal CRISPRmap REPEATS data that was used for the clustering?
The original REPEATS data set in a single fasta file and
the species acronyms used for the IDs can be downloaded here:
