
Introduction
CRISPRmap provides a quick and detailed insight into repeat conservation and diversity of both bacterial and archaeal systems. It comprises the largest dataset of CRISPRs to date and enables comprehensive independent clustering analyses to determine conserved sequence families, potential structure motifs for endoribonucleases, and evolutionary relationships.
When using CRISPRmap please cite :
- Omer S. Alkhnbashi, Fabrizio Costa, Shiraz A. Shah, Roger A. Garrett, Sita J. Saunders, and Rolf Backofen.
CRISPRstrand: predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci
Bioinformatics, 2014, 30(17), 489-496. - Sita J. Lange, Omer S. Alkhnbashi, Dominic Rose, Sebastian Will and Rolf Backofen.
CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems
Nucleic Acids Research, 2013, 41(17), 8034-8044. - Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.
Results are computed with CRISPRmap version meta
Overview
The following parameters are used to control the execution of CRISPRmap
Furthermore, additional information is available
Input Parameters
 Sequence Input in FASTA Format
Sequence Input in FASTA Format
CRISPRmap accepts input in the form of one or more CRISPR sequences
(repeat sequence only) in FASTA format.
For a CRISPR array, a single single repeat sequence should
be chosen that is either most common or represents the consensus of
all repeat instances in the array.
You can choose between directly typing in (or pasting) your FASTA formatted sequences into the text field or
uploading a text file containing the FASTA formatted sequences to the web server.
A FASTA sequence entry needs to have a description line (also called header line, starting with “>”),
typically including a sequence ID and optionally a description, followed by the corresponding sequence in a new line.
A simple example for illustration:
>Repeat1 GTGCTCAACGCCTTACGGCATCAATGGTTTGGACAC >Repeat2 GTTTTAAATCAGTTAATTTCTCCTACGAGTCGAGAC >Repeat3 ATGTTCCCCACACGTGTGGGGATGAACCGNote that the sequence ID you choose will later be the user ID that identifies your sequence (along with an assigned ID for calculation) in the results, so you might want to consider using a unique sequence ID name. Also, only the first word in the header line (all characters up to the first space character in the line) will be used as the user ID. Moreover, if the word extends 13 characters, only the first 13 characters will be used. Currently, a maximum of 400 CRISPRs are allowed as an input.
The parameter constraints are: The input has to be in valid FASTA format. No multiline input is allowed (sequence etc. in one line each). The number of sequences has to be at least 1 and at most 400. Sequence lengths have to be in the range 1-50. The allowed sequence alphabet is 'ACGUTacgut'.
Defaults to ()
Defaults to ()
Optimize reading direction of input sequences
If you are not sure about the correct repeat sequence orientation (i.e. you don't know whether you have chosen the right strand)
you can enable this option and allow CRISPRmap to predict the orientation to make it consistent with our data (based on a machine learning prediction model, see manuscript).
Note that regardless of choosing this option, or not, CRISPRmap will check both directions of the given input repeat sequences
for their occurrence in our database. We require a 100 % match to one of the consensus repeat sequences
(from all repeat instances in a CRISPR array) in our dataset in order for the given input
sequence to be reported as occuring in our data. Although slight deviations in consensus sequences
lead to the CRISPR not being found in our dataset, this does not matter, as the sequence will still be clustered
correctly into the CRISPRmap classification system (i.e. will be assigned to correct sequence families and structure motifs).
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Defaults to (false)
Data Set
CRISPRmap version
Defines which version of CRISPRmap is to be used for the prediction:
CRISPRmap v1.*-2013 : contains 3527 consensus repeats, where a group of 40 conserved repeat sequence families were identified together with a total of 33 potential structural motifs. For further details, please refer to our CRISPRmap article 2013.
CRISPRmap v2.*-2014 : increased CRISPR data set (4719 consensus repeats), where a group of 24 conserved repeat sequence families are identified together with a total of 18 potential structural motifs. Due to the new class definition the number of conserved repeat sequence families and structural motifs are decreased. Additionally, CRISPRstrand is integrated in CRISPRmap and it uses the most recent classification of Cas subtype. For further details, please refer to our CRISPRmap article 2014.
CRISPRmap v1.*-2013 : contains 3527 consensus repeats, where a group of 40 conserved repeat sequence families were identified together with a total of 33 potential structural motifs. For further details, please refer to our CRISPRmap article 2013.
CRISPRmap v2.*-2014 : increased CRISPR data set (4719 consensus repeats), where a group of 24 conserved repeat sequence families are identified together with a total of 18 potential structural motifs. Due to the new class definition the number of conserved repeat sequence families and structural motifs are decreased. Additionally, CRISPRstrand is integrated in CRISPRmap and it uses the most recent classification of Cas subtype. For further details, please refer to our CRISPRmap article 2014.
Output Description
Following job completion,
CRISPRmap automatically directs you to the output page,
which contains the clustering results for all the input sequences.
The page is basically divided into four main sections from top to bottom:
CRISPRmap tree of consensus CRISPR sequences:
The CRISPRmap tree, right on the top of the
output page, contains all the consensus CRISPR sequences in the
database clustered together with the given input sequences,
whose positions in the tree are marked with red lines that intersect the
coloured annotation rings around the cluster tree. It basically displays a map
of sequence and structure conservation of all the database members, together with the
input sequences (which are either part of the database or not).
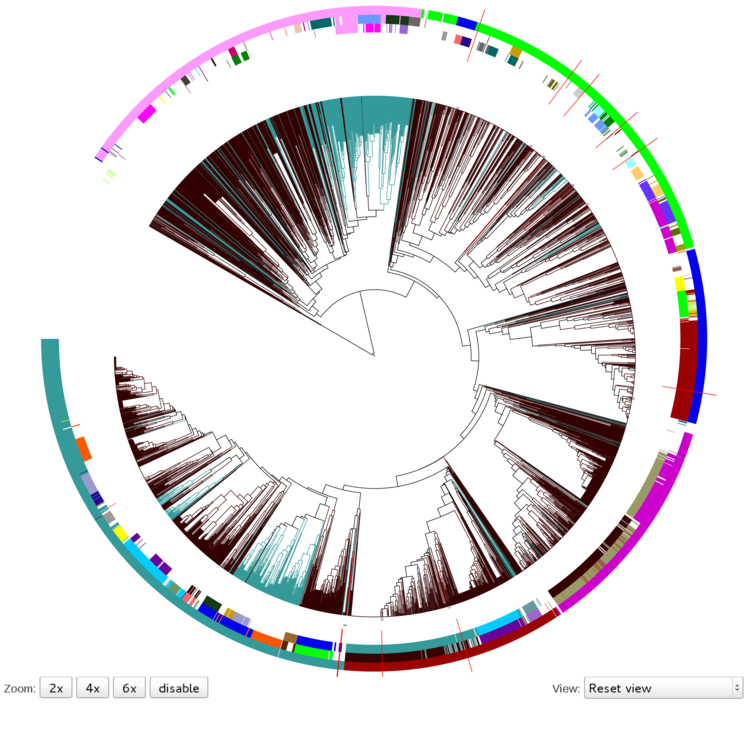
As you can see in the figure above, there are three annotation rings
around the hierarchical tree in the default view (Reset view). The rings from in- to outside
correspond to the following annotations:
structure motif, sequence family and superclass. This means that if you are looking at a
certain position in the tree (e.g. one of the red lines that mark the clustered input repeats),
you can easily tell to which structure motif, sequence family or superclass it is assigned to by
just looking at the underlying colours of the rings and then look up the colour-coding in the legend.
Here’s an example.

Let's look at these two red lines (using the zoom function) that mark the
positions of two clustered input repeats in the tree. On top we can see that they fall in between the
brown tree branches (meaning that they are clustered into the Bacteria domain, opposed to the blue-green
branches marking the Archaea domain) and that they share the same annotation ring colours. To get the class
assignments for the two repeats, we just have to look up the colours in the legend:

For the structure motif (inner ring) we get structure motif 1 (blue-green),
for sequence family and superclass (two outer rings) we get sequence family 2 (brown)
and superclass B (red) for both repeats. You can also find this information in the
“Class assignments” section (below the overview map section) for each input repeat sequence.
Note that additional annotation rings can be selected via the View pop-down window. Other available annotation rings are:
(1) “Cas subtypes combined” with additional rings for both Cas subtype annotations from Haft et al. 2005
(third ring) and Makarova et al. 2011 (fourth ring) (see manuscript for references);
(2) “Taxonomic phyla” with the third ring being the phyla that the Organism belongs to from which the CRISPR was taken;
and (3) “Altogether” contains all annotations – structure motifs (ring 1), sequence families (ring 2),
Cas subtypes Haft 2005 (ring 3), Cas subtypes Makarova 2011 (ring 4), phyla (ring 5), and superclass (ring 6).
Class assignments:

The class assignments table shows you the assigned structure motif,
the sequence family and the superclass for each of the user-given repeat sequences marked by red lines in the
CRISPRmap tree. By clicking on the respective structure motif or sequence family link, you can directly go to the
corresponding sections and see all the structure motif
and sequence family details.
An input sequence can either be identified as a database member or a “new” sequence
(i.e. not part of the database).
CASE 1: if an input sequence is not part of the database,
it will be marked in green and bold. Note that this can also happen if the sequence is
only slightly different from the corresponding consensus CRISPR repeat in our database,
because we require a sequence to match 100 % to a database member in order for it to be identified.
Thus, assigning the feature “new” to a sequence does not necessarily mean that it is unknown or has no corresponding
database sequences. However, this is not a problem for a correct classification, since the sequence will be classified
into the correct cluster anyway. Here’s an example of the input description when an input sequence is classified as new (ID 1 to 9 in the above table):
The first ID (ID 1 to 9) of the input sequence is assigned by CRISPRmap automatically and it serves as a unique identifier on the results page.
The second ID (for example UserId=AcaccL30) denotes the user supplied ID (AcaccL30) for the input sequence.
CASE 2: if an input sequence is identified as a database member
(for example ID 10 UserId=CjeikL29 CRISPRmap=Cjei_A_G_1_M1_F2 in the above table),
we get an input description that features some additional information: The first ID is again the internal CRISPRmap ID,
followed by the second user supplied ID. Afterwards, a compact description of the repeat is given: species abbreviation (CjeikL29),
strain abbreviation (A), genetic element from which repeat originates (G: chromosome, P: plasmid),
motif and family (M1_F2, or X_X if unassigned).
If no structure motif or sequence family could be assigned to the input sequence, a dash is printed
instead of the assigned structure motif or sequence family (for example ID 4 and 5 in the above table).
This corresponds to the blank spaces in the CRISPRmap tree above.
Close up on structure motifs:
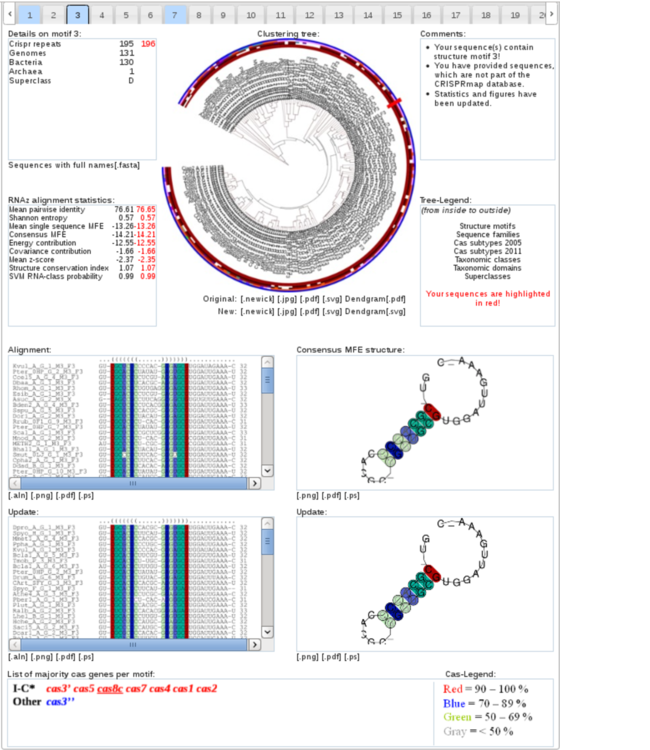
This section offers you comprehensive information on all 33 structure motifs.
The tabs on top of the section allow for quick access to each of the 33 structure motifs and appear in two different colours.
A blue colour indicates that the respective structure motif contains one or more of the input repeats,
while grey-coloured tabs highlight structure motifs with no assigned input repeat sequence. Additionally,
the drop-down box to the left can be used to select a certain structure motif.
The clustering tree for the selected structure motif is presented in the middle. In case of a blue tab,
it also contains the input repeats that feature the motif.
These input repeats can be marked by a red rectangle.
If a new repeat sequence is present, the original (without the new repeat) and the updated cluster tree
(containing the clustered new repeat) can be downloaded right underneath it.
The cluster tree uses the same ring annotation as in the CRISPRmap cluster tree above (“Alltogether” view).
Just like in the CRIPRmap tree, if an input sequence is not in the database
(classified as new) no Cas subtypes or taxonomy can be assigned to it
(blank colour annotation).
Below the tree, a multiple sequence alignment (MSA) of the structure motif,
as well as the consensus minimum free energy (MFE) structure, both calculated by LocARNA, are given
(LocARNA colour-coding described at the
LocARNA help page).
If a new repeat is assigned to the depicted structure motif and this changes the original structure motif,
then we show both the old and new structure motifs (with and without the input repeat sequence(s)).
On the bottom of the structure motif section, a cas genes are listed that are found in the majority of genomes (more specifically only the chromosomes or plasmids)
from which the CRISPRs of the respective motif were extracted.
The cas genes are coloured according to how many genomes they occur in:
less than 50 % (grey), 50-69 % (green), 70-89 % (blue), and above 90 % (red).
The cas genes are further categorised according to the Cas subtypes from Makarova 2011.
In particular, if cas genes were present that were unique to a specific subtype,
then this subtype is listed in one row. Unique genes are underlined.
Nearly complete Cas subtypes are indicated by a star. Ambiguous cas genes (that could belong to more than one subtype) are listed in the final “Other” row.
Close up on sequence families:

Here you can find detailed information on all 40 sequence families. Analogous to the
“Close up on structure motifs” section above,
we again have a drop-box down to the left and tabs on top for
quick access that are either blue for sequence families with assigned input repeats or grey for
sequence families with no assigned input repeats.
We use the WebLogo tool (http://weblogo.berkeley.edu)
to generate sequence logos of all aligned family members.
Sequence logos present a graphical representation of a multiple sequence alignment,
in this case of the family member repeat sequences. Again, if an input sequence
is assigned to the respective sequence family and it results in some
changes, the old (without the input sequence(s)) and the new (with input sequence(s))
results are depicted.
Downloads
Here you can download all the job results packed into one .zip archive file,
including all the pictures, tables and files downloadable in the separate sections.
Additionally, you can download a table with annotation information for all cas genes in
the database in .tab (values separated by tabs) format.
The original REPEATS data set in a single fasta file and the species acronyms used for the IDs can be downloaded here:
The original REPEATS data set in a single fasta file and the species acronyms used for the IDs can be downloaded here:
Input Examples
CRISPRmap for 8 repeats (v2.1)
CRISPRmap for 8 repeats (v2.1)
The example's result can be directly accessed here
CRISPRmap for 16 repeats (v1.0)
Example for version 1.0 and 16 repeats
The example's result can be directly accessed here
Frequently Asked Questions
If your question is not listed, please send it to us!
 Where can I download the orginal CRISPRmap REPEATS data that was used for the clustering?
Where can I download the orginal CRISPRmap REPEATS data that was used for the clustering?
The original REPEATS data set in a single fasta file and
the species acronyms used for the IDs can be downloaded here:
List of Changes
- 4.0.5 : CRISPRmap v2.1.3 and v1.3.0 online
- 3.4.2 : CRISPRmap 2.1 online
- 3.3.0 : CRISPRmap 2.0 online : increased CRISPR data; enhanced CAS-subtype annotation and new annotation for archea; new class definition
- 3.0.0 : CRISPRmap 1.0 online
About this web server
|
version 5.0.12
|
Copyright © 2012 - 2025 Bioinformatics Group Freiburg
|
 |
Imprint and Disclaimer
|
Imprint and Disclaimer