Introduction
LocARNA is a tool for multiple alignment of RNA molecules.
It is one of the fastest and most accurate available tools that do a
true sequence and structure based comparison of the RNAs. LocARNA
requires only RNA sequences as input and will simultaneously fold and
align the input sequences. LocARNA outputs a multiple alignment together
with a consensus structure. Thus LocARNA is comparable with methods like
Dynalign, Foldalign, and Lara.
For the folding it makes use of a very realistic energy model for RNAs
which is also employed by RNAfold of the Vienna RNA package (or Zuker's
mfold). For the alignment it features RIBOSUM-like
similarity scoring and realistic gap cost.
The high performance of LocARNA is mainly achieved by employing base pair
probabilities during the alignment procedure. This allows to sort out
insignificant base pairs from the start. Since the base pair probabilities
stem from a full energy model, LocARNA maintains the high accuracy of
Sankoff-like RNA comparison methods.
The LocARNA software is available for download as part of the
LocARNA
package (GPL 3). In contrast to the server, the stand-alone software
does not limit the problem size, provides enhanced functionality, and
offers a batch processing-friendly command line interface.
To get a first idea and an impression of LocARNA's abilities, please
check out our
LocARNA at RNA-tools Freiburg
tutorial video (older webserver version):
When using LocARNA please cite :
- Sebastian Will, Tejal Joshi, Ivo L. Hofacker, Peter F. Stadler, and Rolf Backofen.
LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs
RNA, 18 no. 5, pp. 900-14, 2012
- Sebastian Will, Kristin Reiche, Ivo L. Hofacker, Peter F. Stadler, and Rolf Backofen.
Inferring non-coding RNA families and classes by means of genome-scale structure-based clustering
PLoS Computational Biology, 3 no. 4, pp. e65, 2007
- Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.
Results are computed with LocARNA version 1.9.1 linking Vienna RNA package 2.3.2
Overview
The following parameters are used to control the execution of LocARNA
Furthermore, additional information is available
Sequence and Structure
 Sequence Input in FASTA Format
Sequence Input in FASTA Format
LocARNA accepts input in form of a multiple FASTA file. An example looks like this:
>fruA
CCUCGAGGGGAACCCGAAAGGGACCCGAGAGG
>fdhA
CGCCACCCUGCAACCCAAUAUAAAAUAAUACAAGGGAGCAGGUGGCG
>vhuU
AGCUCACAACCGAACCCAUUUGGGAGGUUGUGAGCU
>hdrA
GGCACCACUCGAAGGCUAAGCCAAAGUGGUGCU
Input can be given either as direct text input or uploading a file.
Note: Input size is limited to restrict computation time and memory requirements.
Anchor and Structure Constraints
Along with the input sequences, one can specify constraints on the
alignment, including anchor constraints as well as structure
constraints.
The
structure constraints (lines #S in the following example)
inherit their semantics from RNAfold:
- . = no constraint at all
- x = base must not pair
- ( ) = matching brackets encode base i pairs base j
- | = paired with another base
- > = base i is paired with a base j>i
- < = base i is paired with a base j<i
In consequence, the alignment can only be guided by base
pair matches that are compatible to the given constraints.
Structure constraints can be specified for individual sequences.
>fruA
CCUCGAGGGGAACCCGAAAGGGACCCGAGAGG
.......(((..(((xxxx))).)))...... #S
>fdhA
CGCCACCCUGCGAACCCAAUAUAAAAUAAUACAAGGGAGCAGGUGGCG
..............(((.....xxxxxx......)))........... #S
Note that for input RNAs with structure constraints (#S or #FS), we
automatically fall back to non-local folding (unless the user chooses to
ignore all constraints).
The
anchor
constraints are specified by giving unique names to certain sequence
positions, here A1,A2,A3,A4,A5,A6,B1,B2,B3,C1,C2,C3,C4 (lines
#1,#2). Positions of the same name in different sequences are
aligned. In each sequence, names have to be unique.
Note: anchor constraint lines have to be specified
for none or all sequences,
but they can be empty (only dots) for individual sequences!
>fruA
CCUCGAGGGGAACCCGAAAGGGACCCGAGAGG
.........AAAAAA.BBBCCCC......... #1
.........123456.1231234......... #2
>fdhA
CGCCACCCUGCGAACCCAAUAUAAAAUAAUACAAGGGAGCAGGUGGCG
...........AAAAAA.....BBB.........CCCC.......... #1
...........123456.....123.........1234.......... #2
Fixed Structures
Instead of structure constraints (lines #S) you can also specify fixed structures using lines #FS as follows:
>fruA
CCUCGAGGGGAACCCGAAAGGGACCCGAGAGG
((((..(((...(((....))).)))..)))) #FS
>fdhA
CGCCACCCUGCGAACCCAAUAUAAAAUAAUACAAGGGAGCAGGUGGCG
(((((((.(((...(((.................))).)))))))))) #FS
Whereas structure constraints (#S) only specify parts of the structure and are used to create dot plots representing the ensemble of all structures being compatible with the constraints, fixed structures (#FS) force the ensemble considered for this sequence to contain only this one, fixed structure.
The #FS string can contain pseudoknots; for this purpose, LocARNA supports various bracket symbols: (),[],{},Aa,Bb,Cc,Dd.
Sequences without any given structure, sequences with structure constraints (#S) and sequences with fixed structures (#FS) can be mixed freely.
Note that for input RNAs with structure constraints (#S or #FS), we
automatically fall back to non-local folding (unless the user chooses to
ignore all constraints).
The parameter constraints are: The input has to be in valid FASTA format. The number of sequences has to be at least 2 and at most 30. Sequence lengths have to be in the range 5-2000. The allowed sequence alphabet is 'ACGUTNacgutn'. Fixed structure can be given in a single line with tailing '#FS' using the brace pairs ()[]{}AaBbCcDd. Structure constraints can be given in a single line with tailing '#S' using the alphabet ().x|<>. Anchor constraints can be given. In LocARNA-P (probabilistic) mode, the maximal sequence length is 1000nt.
Defaults to ()
Alignment
Alignment Type
Perform either global or local alignment. In global mode, the whole input sequences are aligned, whereas local alignment will determine the best matching subsequences of the input sequences.
Alignment Mode
In standard mode,
the server runs LocARNA's core engine. For most needs,
this mode provides a good balance of alignment accuracy and efficiency.
In LocARNA-P mode,
the server delegates the alignment to the tool
LocARNA-P. The latter annotates the alignment with a reliability
profile; this versatile information enables you to analyze the local
alignment quality, revealing conserved sequence and structure signals in
your sequences. Furthermore, employing consistency transformation,
LocARNA-P usually bests LocARNA's alignment accuracy.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to ( Standard)
Alignment Scoring
Structure Weight
Weights structural match against sequence match and gap cost. A structural match of two arcs is assigned a score of at most 2. The default structure weight of 200 turned out to balance well the score contributions of structure match and sequence alignment.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0 and must be smaller than or equal to 2000.
Defaults to (200)
Indel Opening Score
Score for starting a gap in the alignment. This score is a penalty and therefore should be negative.
The parameter constraints are: Input value has to be parsable as Integer. The value must be smaller than or equal to 0.
Defaults to (-800)
Indel Score
Cost of extending an alignment gap.
The parameter constraints are: Input value has to be parsable as Integer. The value must be smaller than or equal to 0.
Defaults to (-50)
Use RIBOSUM
Whether or not the RIBOSUM matrix 'RIBOSUM85_60' is to be used for scoring sequence match/mismatch. RIBOSUM scoring is the default for LocARNA. If one disables the RIBOSUM matrix use, sequence matchs/mismatchs are scored as given explicitely by parameters 'Match Score' and 'Mismatch Score'.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
Match Score
Score for aligning two identical nucleotides.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0.
Defaults to (50)
Mismatch Score
Score for aligning two different nucleotides.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0.
Defaults to (0)
RNA Folding
Local folding: max. base pair span
If set, local folding will be applied which limits the
maximal span width of a base pair to the given number of
nucleotides (nt)
(see '-L' in
RNAplfold manual).
Local folding is key to reasonable folding results when facing
larger RNA molecules, since it minimizes effects of incorrect
long-range predictions (see
local folding article).
Note, setting the span width automatically adjusts the folding
window size
(see '-W' in
RNAplfold manual).
to
2*span!
Note further that for input RNAs with structure constraints (#S or #FS), we
automatically fall back to non-local folding (unless the user chooses to
ignore all constraints).
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than 2.
Defaults to (150)
Temperature in °C
Rescales the RNA folding energy parameters to the given temperature
in degree Celsius (°C)
(see '-T' in
RNAfold manual).
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 0 and must be smaller than 100.
Defaults to (37)
Energy parameter set (Vienna package)
Defines what energy parameter set to be used within
the Vienna RNA package to compute base pair probabilities,
i.e. dot plots, for each input sequence. The parameter sets are
provided by the Vienna RNA package (see version information) and are
named according to the first author and year of the corresponding
publication.
Upload dot plots
By default, LocARNA creates a dot plot (base pair probability matrix) for each sequence of the fasta input. The dot plot is generated using the tool RNAfold, unless you specify a fixed structure. Alternatively, you can upload custom dot plots; these dot plots are specified in the Vienna RNA dot plot format as it is generated by RNAfold (post script, .ps, please see RNAfold man page). To specify the dot plot of a particular sequence in the fasta input, the sequence in the uploaded file has to exactly match that sequence in the fasta input; file names and the order of uploads are not relevant there. It is possible to upload dot plots for only some of the sequences; then, LocARNA will still compute dot plots for the remaining sequences.
Heuristics for speed/accuracy tradeoff
Minimal Pair Probability
Only base pairs that have at least the minimal pair probability are considered for scoring the alignment. Base pairs with lower probability are considered insignificant.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 2.0E-4 and must be smaller than 1.
Defaults to (0.0005)
Minimal Probability for Guide Tree Construction
Same as minimal pair probability but only used for alignments during the construction phase of the guide tree. Since those alignments are less important than alignment during the progressive alignment phase, one can use higher values here than for "Minimal Pair Probability".
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 0 and must be smaller than 1.
Defaults to (0.005)
Maximal Difference for Sizes of Matched Arcs
Restrict the length difference of base pairs that can be matched. A value of '-1' disables this restriction.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to -1 and must not be equal to 0.
Defaults to (30)
Maximal Difference for Alignment Edges
Restrict the difference of sequence positions that can be matched. A value of '-1' disables this restriction.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to -1 and must not be equal to 0. If not -1, it has to hold (maxSequenceLength > (maxDiff * 2 * (minSequenceLength-1))).
Defaults to (60)
Other Parameters
Ignore Constraints
Ignore anchor constraints and structural constraints, if they are specified in the input. Otherwise this option has no effect.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Disallow Lonely Pairs
Forbid the occurence of lonely base pairs in the consensus structure of an alignment.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
Use alifold consensus dot plots
Employs RNAalifold -p for generating the consensus dot plot after each
progressive alignment step. Otherwise, the consensus
dot plot are computed as averages over the input dot plots.
Note: This parameter can not be applied and is
automatically disabled if (a) structure constraints (#S or #FS) are
provided, (b) folding temperature and energy parameters are changed,
or (c) LocARNA-P computations are requested.
The parameter constraints are: Input value has to be parsable as Boolean. Is automatically disabled in case Temperature in °C and Energy parameter set (Vienna package) differ from their standard values. Furthermore, it can not be applied if structure constraints are present or LocARNA-P computations are requested.
Defaults to (false)
Consistency Transformation (LocARNA-P only)
Consistency-transformation improves the multiple alignment accuracy by avoiding mistakes during progressive aligment. It will re-estimate match probabilities in the multiple alignment based on pairwise alignment probabilities in sequence triples.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Keep Sequence Input Order
If enabled, the original sequence input order will be preserved
within the generated alignment.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Stockholm alignment output
If enabled, the all alignments are also produced in Stockholm format.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
Output Description
The alignment is annotated with its consensus structure, a secondary structure of the alignment, as it is predicted by RNAalifold
The consensus structure is printed as a string of dots and brackets on top of the alignment. The string is well-bracketed, such that base pairs in the structure are
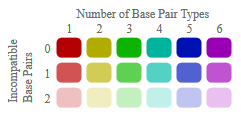
indicated by corresponding opening and closing brackets. Furthermore, compatible base pairs are colored, where the hue shows the number of different types C-G, G-C, A-U, U-A, G-U or U-G
of compatible base pairs in the corresponding columns. In this way the hue shows sequence conservation of the base pair. The saturation decreases with the number of incompatible base pairs.
Thus, it indicates the structural conservation of the base pair
The representation was generated by the tool RNAalifold of the Vienna Package.
Furthermore, we provide file downloads of the alignment information in the following formats:
- [ClustalW] - a standard alignment format of the aligned sequences only
- [FASTA] - a standard sequence format, which is extended with the consensus structure in the first line (non-standard)
- [structure] - the consensus structure only in common dot-bracket notation
- [structure-bp] - position pairs for the base pairs in the consensus structure with indices starting with 1. This is e.g. needed when using the structure information in Mr.Bayes NEXUS format.
Visualization of the local sequence–structure-based alignment
reliability (STAR) of the result alignment. The profile consists of the
reliabilities for each single alignment column. The dark regions
indicate structure reliability, the light regions represent sequence
reliability, and the thin line shows the combined column-reliability.
The column-wise reliabilities are computed as sum-of-pairs over match
probabilities, which are computed by LocARNA-P.
The guide tree reflects the similarity relations between your input
sequences in the form of a hierarchical clustering. This tree is
computed by LocARNA to guide its multiple alignment. It is constructed
by the UPGMA method based on the all-against-all comparison of the
sequences by LocARNA; the latter considering sequence and structure
similarity. The guide tree view is generated using the
Newick Utilities.
The consensus structure of the alignment, as predicted by RNAalifold, is shown in a 2D layout.
Base pairs are colored using the same color code as in "Colour and structure annotated alignment", such that hue shows sequence conservation and saturation shows structural conservation.
Per alignment position, the set of nucleotides with frequency higher than average are represented using
IUPAC ambiguity codes for nucleotides.
If gaps are present, lower letters are used.
If the gap symbol is most frequent, a gap symbol is shown.
The representation was generated by the tool RNAalifold of the Vienna Package.
Compatible base pairs are colored, where the hue shows the number of different types C-G, G-C, A-U, U-A, G-U or U-G
of compatible base pairs in the corresponding columns. In this way the hue shows sequence conservation of the base pair. The saturation decreases with the number of incompatible base pairs.
Thus, it indicates the structural conservation of the base pair.
Input Examples
snoRNAs with constraints
snoRNAs with constraints
The
example's result can be directly accessed
here tRNA alignment with fixed structure
tRNA alignment with fixed structure
The
example's result can be directly accessed
here RNA Boundaries with LocARNA-P
RNA Boundaries with LocARNA-P
The
example's result can be directly accessed
here tRNA alignment
tRNA alignment
The
example's result can be directly accessed
hereFrequently Asked Questions
If your question is not listed, please send it to us!
 is faster, why should I use LocARNA for aligning RNAs?") ClustalW (or my favourite sequence alignment tool) is faster, why should I use LocARNA for aligning RNAs?
ClustalW (or my favourite sequence alignment tool) is faster, why should I use LocARNA for aligning RNAs?
Sequence alignment programs like ClustalW, T-Coffee, MUSCLE, or MAFFT compare RNAs only by sequence similarity. However, for many RNAs the structure information is strongly conserved. Thus, sequence similarity can be weak even for closely related RNAs of the same family. Consequently, only sequence-structure alignment programs that consider sequence and structure similarity, such as LocARNA, can reveal their true similarity. Pure sequence alignment programs will usually completely fail if sequence identity drops below 60%. Unfortunately, fully taking structure information into account is computationally expensive. Nevertheless, LocARNA achieves very good performance for this class of algorithms.
Why should I use LocARNA compared to the RNA alignment program XY?
LocARNA is one of the fastest programs that do true sequence-structure alignment of RNAs and therefore produce highly accurate alignments. The LocARNA server is nice and offers a rich interface. You can even specify anchor constraints and structure constraints for global and local alignment; also probabilistic multiple alignment with alignment reliabilities is unique to LocARNA. Honestly, there are other very good programs out there; try them and compare!
How does LocARNA achieve its speed and low space consumption? Does this compromise accuracy?
LocARNA uses different heuristics for improving speed and lowering space requirements. Most importantly, it filters base pairs by their probability in the RNA structure ensemble and considers only 'significant' base pairs that pass a certain probability threshold. This reduces complexity from O(n^6) time and O(n^4) space to O(n^4) time and O(n^2) space, respectively. Furthermore, one can control which base pairs are compared at all by setting a maximal length difference. Finally, as reasonable for global alignment, one can control which residues are compared by LocARNA by a maximal difference of sequence positions. All those heuristics are optional and can be controlled in the advanced section of the server. However, the heuristics with default settings were shown to preserve alignment accuracy on the comprehensive Bralibase 2.1 benchmark. It's therefore likely that default settings won't compromise the alignment accuracy for your RNAs.
I am using LocARNA in local alignment mode; why does LocARNA return an empty multiple alignment (or only a very small one)?
Generally, LocARNA constructs multiple alignments by progressively merging (previously computed) alignments of fewer sequences. In the case of local alignment, LocARNA merges local subalignments from previous progressive alignment steps. In this way, the alignments can become shorter and shorter with every merge, since each progressive step retains only the sufficiently similar parts of the subsequences. Thus, if LocARNA cannot not find sufficiently similar common subsequences in all of your input sequences, it will produce a very small or even empty alignment.
Even in the latter case, LocARNA still computes the relations between all of your sequences in the form of a guide tree; furthermore it computes the alignments of the sequence subsets corresponding to the inner nodes of this tree. In many cases, one is rather interested in those alignments than in one single alignment of all sequences.
I want to align more sequences than allowed. What can I do?
Often it is a good idea to first look for high sequence similarity in
such a set of sequences. If one can identify sequence pairs with e.g.
more than 80% or 90% identity, usually it does not pay off to align them based
on structure similarity. Please check this by running a multiple
sequence alignment first.
If this is the case with your set of sequences, you could reasonably
reduce the number of sequences that you feed to our web server by
omitting extremely similar sequences.
I want to align longer sequences than allowed. What can I do?
An idea that might work: split the long sequence into (e.g. three)
overlapping "windows", i.e. subsequences, and use the web server.
This is especially useful, if your are aligning short sequences with
one (or few) long sequence(s) looking for a conserved motif.
RNAalifold consensus structure output shows single-letter codes, what is the meaning?
The consensus structure output is produced with RNAalifold's option '--mis', i.e. RNAalifold
outputs the "most informative sequence" instead of simple consensus: For each
column of the alignment output the set of nucleotides with frequence
greater than average in IUPAC notation
(see
RNAalifold manual page).
This means that the special letters are
IUPAC codes for nucleotides.
This is especially useful, if your are aligning short sequences with
one (or few) long sequence(s) looking for a conserved motif.
List of Changes
- 4.4.4 : default values changed: gap opening -800 (was -500) and gap extension -50 (was -350)
- 4.4.2 : consensus structure fixed; alignment output fixed; check in probabilistic mode: max length difference less than 200
- 4.4.0 : LocaARNA v1.9.1 online
- 4.3.0 : LocARNA v1.8.11 online linking Vienna RNA v2.2.7: bugfix for long sequences
- 4.2.3 : linking Vienna RNA bugfix release 2.2.7; NEW: alignment in Stockholm format for download
- 4.2.2 : LocARNA v1.8.10 online: long name handling improved, anchor constraint handling fixed, ..
- 4.0.8 : LocARNA v1.8.5 online: minor bugfixes
- 3.4.0 : LocARNA v1.8.0 online: new advanced options to control RNA folding
(temperature, energy parameters, local folding); by default, long RNAs
are now folded locally; fixed structure input example
- 3.3.6 : optional input order preservation for output enabled
- 3.3.1 : bugfix in postprocessing for structure plot generation
- 3.3.0 : LocARNA v1.7.16 online : bugfix concerning iterative alignment and alignment evaluation; alignment and consensus structure available for download
- 3.2.3 : maximal sequence length in LocARNA-P mode now 1000nt
- 3.2.0 : LocARNA v1.7.13 online : bugfix for structure constraint usage
- 3.1.5 : LocARNA v1.7.11 online : Improved max-diff heuristic (in case of sequences of unequal length.)
Due to this change, 1) max-diff works strictly symmetric, i.e. it
behaves the same whether aligning A vs B or B vs A, and 2) the heuristic
can not fail anymore, since too small values are increased automatically.
- 3.1.2 : LocARNA v1.7.10 online
- 3.1.1 : post-processing bugfix
- 3.1.0 : LocARNA v1.7.9 online : local progressive alignment on subalignments; guide tree visualization
- 2.7.7 : LocARNA v1.7.7.1 online
- 2.7.6 : additional examples