
AntaRNA is an Ant-Colony Optimization based tool which solves the RNA inverse
folding problem. It designs RNA sequences which satisfy a set of
constraints made by the user. The realized multi-objective optimization allows
to introduce structure, sequence and GC-content constraints.
The multi-objective optimization is modeled on different layers
within a terrain-graph on which simulated ants explore pathes. During a walk of an ant, it
assembles a sequence based on the information that is stored in the graph.
Dependent on the quality of the solution, parts of the graph, which are in compliance with the constraints,
get promoted, such that their selection
probability increases.
Subsequent ants produce and evaluate further solutions and modify the terrain
graph accordingly until a solution sequence was found, which satisfies the
user defined constraint set.
Introduction
When using AntaRNA please cite :
- Robert Kleinkauf, Martin Mann and Rolf Backofen
AntaRNA - Ant Colony Based RNA Sequence Design
Bioinformatics, 31(19), pages 3114-3121, 2015. - Robert Kleinkauf, Torsten Houwaart, Rolf Backofen, and Martin Mann
AntaRNA - multi-objective inverse folding of pseudoknot RNA using ant-colony optimization
BMC Bioinformatics, 16(1), pages 1-7, 2015. - Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.
Results are computed with AntaRNA version 1.1.2 using Vienna RNA package 2.4.14 or pKiss 2.2.14
Overview
The following parameters are used to control the execution of AntaRNA
Furthermore, additional information is available
Constraints
 Structure constraint
Structure constraint
The RNA secondary structure, you wish to design a sequence for, has to be given in extended bracket notation.
A base pair between bases i and j is represented by a '(' at the ith position and a ')' at position j.
Unpaired bases are represented by dots.
If pseudoknots are to be encoded, you can use the brace pairs (), [], {}, <>.
Example:
The following structure is represented by the string (((.((.(((....))))).))).
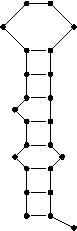
Besides the regular dot bracket structure notation, AntaRNA provides the usage of 'fuzzy' structure constraints. Respectively defined regions within a structure are allowed to form any structure which occurs, as long as the occurring structure is only interacting within the defined block of constraint.
Using soft constraint (lower case letters) mode, no structure is enforced within the blocks, whereas in hard constraint (upper case letters), at least one base pair has to form within such a defined block. The blocks are allowed to be defined by regular letters and include : "ABCDEFGHIJKLMNOPQSTUVWXYZabcdefghijklmnopqrstuvwxyz"
A base pair between bases i and j is represented by a '(' at the ith position and a ')' at position j.
Unpaired bases are represented by dots.
If pseudoknots are to be encoded, you can use the brace pairs (), [], {}, <>.
Example:
The following structure is represented by the string (((.((.(((....))))).))).
Besides the regular dot bracket structure notation, AntaRNA provides the usage of 'fuzzy' structure constraints. Respectively defined regions within a structure are allowed to form any structure which occurs, as long as the occurring structure is only interacting within the defined block of constraint.
Using soft constraint (lower case letters) mode, no structure is enforced within the blocks, whereas in hard constraint (upper case letters), at least one base pair has to form within such a defined block. The blocks are allowed to be defined by regular letters and include : "ABCDEFGHIJKLMNOPQSTUVWXYZabcdefghijklmnopqrstuvwxyz"
The parameter constraints are: Has to be either a balanced nested structure encoded by brackets '()'or a balanced crossing pseudoknot structure using the brace pairs '()[]{}<>'. Base pairs have to have a minimal loop length of 3. Positions that have to be unpaired are encoded by '.'. Implicit structure constraints use the alphabet 'ABCDEFGHIJKLMNOPQRSTUVWXYZabcdefghijklmnopqrstuvwxyz' where upper/lower case encode hard/soft block constraints, respectively. String length has to be in range (5,300). Maximally 1 line is allowed.
Defaults to ()
Defaults to ()
Sequence constraint
Sequence constraint using IUPAC ambiguity codes for nucleotides {ACGTURYMKWSBDHVN} with wild-card "N".
A detailed list of the codes is given below.
Note, upper case nucleotide codes are enforced in the designed sequences while lower case codes are 'soft' constraints and only penalized if not present in the solution sequences.
The sequence constraint can also be used as soft constraint. For this, the user can specify lower case characters a,c,g and u. In those cases the terrain is not pruned as in the cases of upper case letters. This allows some rest probabilities for alternative nucleotides. The 'correct' nucleotide is realized via the sequence distance penalty to the quality score of a solution.
Note, upper case nucleotide codes are enforced in the designed sequences while lower case codes are 'soft' constraints and only penalized if not present in the solution sequences.
| IUPAC nucleotide code | Base |
| A | Adenine |
| C | Cytosine |
| G | Guanine |
| U/T | Uracil / Thymine |
| R | A or G |
| Y | C or U/T |
| S | G or C |
| W | A or U/T |
| K | G or U/T |
| M | A or C |
| B | C or G or U/T |
| D | A or G or U/T |
| H | A or C or U/T |
| V | A or C or G |
| N | any base |
The sequence constraint can also be used as soft constraint. For this, the user can specify lower case characters a,c,g and u. In those cases the terrain is not pruned as in the cases of upper case letters. This allows some rest probabilities for alternative nucleotides. The 'correct' nucleotide is realized via the sequence distance penalty to the quality score of a solution.
The parameter constraints are: The IUPAC alphabet 'ACGTURYMKWSBDHVN' (upper case) is allowed for explicit constraint specification and only 'acgtu' (lower case) can be used for soft constraint definitions. If provided, it has to have the same length as the structure constraint. Lower/Upper case symbols represent soft/hard sequence constraints, respectively.
Defaults to ()
Defaults to ()
Targeted GC-content [0..1]
Objective target GC content in [0,1].
This constraint can be extended to uniform or normal GC distributions.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 0 and must be smaller than 1.
Defaults to (0.6)
Defaults to (0.6)
Maximum for uniform distribution sampling
Provides a maximum tGC value [0,1] for the case of uniform GC distribution sampling. The regular tGC value serves as minimum value (tGC < tGCmax).
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 0 and must be smaller than 1. Either this parameter or Variance for normal distribution sampling can be set, not both at the same time.
Defaults to ()
Defaults to ()
Variance for normal distribution sampling
Provides a tGC variance (sigma square) for the case of normal distribution sampling. The regular target GC value serves as expectation value (mu).
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 0 and must be smaller than 1.
Defaults to ()
Defaults to ()
Constraint
Allow GU base pairs
Whether or not GU base pairs are to be considered as valid
base pairs within the structure
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
Defaults to (true)
Allow pseudoknots
Switch to pseudoknot based prediction using pKiss.
The AntaRNA parameters are fixed to optimized values for pKiss.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Defaults to (false)
Energy
Folding temperature
Provides the temperature for the folding algorithm in Celsius
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than 0 and must be smaller than 100.
Defaults to (37)
Defaults to (37)
AntaRNA
Max. terrain resets
Amount of maximal terrain resets, until the best solution is retuned as solution.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 20.
Defaults to (5)
Defaults to (5)
Convergence counts
Delimits the convergence count criterion for a reset.
If the threshold for this counter is exceeded,
the terrain will be rest to its initial state.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 300.
Defaults to (130)
Defaults to (130)
Ant termination criterion
Delimits the amount of internal ants which is allowed to be used until the criterion
'termination convergence' is raised and the program terminates.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 300.
Defaults to (50)
Defaults to (50)
Probability weight alpha
Sets alpha, the probability weight for terrain pheromone influence during the calculation of the edge probabilities.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 1.
Defaults to (1)
Defaults to (1)
Probability weight beta
Sets beta, the probability weight for terrain path influence during the calculation of the edge probability.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 1.
Defaults to (1)
Defaults to (1)
Pheromone evaporation rate
Pheromone evaporation rate. Determines the rate on how much pheromone information evaporates in each round.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 1.
Defaults to (0.2)
Defaults to (0.2)
Constraint weight structure
Structure constraint quality weighting factor.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 10.
Defaults to (0.5)
Defaults to (0.5)
Constraint weight sequence
Sequence constraint quality weighting factor.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 10.
Defaults to (1)
Defaults to (1)
Constraint weight GC-value
GC-value constraint quality weighting factor.
The parameter constraints are: Input value has to be parsable as Double. The value must be greater than or equal to 0 and must be smaller than or equal to 10.
Defaults to (5)
Defaults to (5)
Random seed
Provides a seed value for the used pseudo random number generator.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 99999.
Defaults to ()
Defaults to ()
Output
Number of sequences
Number of sequences which shall be produced.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 1 and must be smaller than or equal to 100.
Defaults to (10)
Defaults to (10)
Output Description
dStr:
The structural distance is based on the
symmetric difference of the base pair sets of the target structure and
the
structure of the current sequence under investigation. The distance is
constraint length normalized.
If the lonely base pair flag is used, the treatment of lonely base pairs
and
2-lonely base pair stacks excludes the explicit structure constraint
during the
structure distance calculation but enforces, that the bases at the
respective
positions potentially could form a base pair. This explains why some
mfe structures
do not show requested base pairs but have a structural distance
of 0.
dSeq:
The sequence distance is realized as an edit distance between the
constraint and
the current sequence. For each time a sequence position does not satisfy
the
provided constraint, the distance grows. The distance is
constraint length normalized.
dGC:
The GC distance of a sequence towards its constraint is simply the
deviation
of the GC content towards the made constraint in its absolute value. For
the
case, in which a certain sequence length cannot achieve a targeted GC
content
explicitly, the next two possible GC values of the sequence are made
legal GC
values within the calculation and thus result in a dGC value of 0.
Input Examples
SECIS block design
Design a SECIS element with flexible stem length (block C) and flanking context (blocks A and B).
Note, where upper case symbols are used the according block has to show at least one base pair in the final design.
The design is visualized in the following figure.
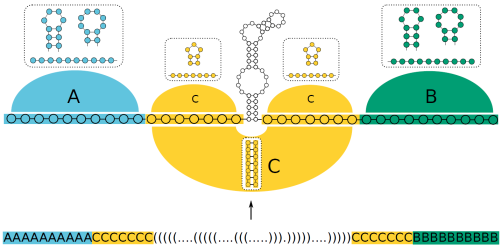
The example's result can be directly accessed here
Crossing pseudoknot
Design sequences for the crossing pseudoknot structures PKB298 with low GC-content.
The example's result can be directly accessed here
Nested tRNA structure
Design sequences for the clover leaf form of a tRNA with high GC-content.
The example's result can be directly accessed here
List of Changes
- 4.4.2 : compatibility check for Cseq and Cstr within the interface
- 4.0.0 : AntaRNA v1.1.2 online
About this web server
|
version 5.0.12
|
Copyright © 2012 - 2025 Bioinformatics Group Freiburg
|
 |
Imprint and Disclaimer
|
Imprint and Disclaimer