

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Use the following button if you want to resubmit the job with
altered input or parameterization:
Input and runtime details for job 7212798 (precomputed example)
Sequence Parameters
 Wild type single RNA sequence in FASTA Wild type single RNA sequence in FASTA | [.fa] | ||
| Mutation encoding | G19U |
Folding options
| Window size (RNAplfold) | 200 | ||
| Maximal base pair span (RNAplfold) | 150 |
Job ID 7212798 (server version trunk)
 | Job Submitted & Queued | @ Tue Apr 14 19:36:55 CEST 2020 | |
| MutaRNA Started | @ Tue Apr 14 19:39:20 CEST 2020 | |
| MutaRNA Finished & Post-Processing | @ Tue Apr 14 19:42:27 CEST 2020 | |
| Post-Processing Finished | @ Tue Apr 14 19:43:53 CEST 2020 | |
| Job Completed | @ Tue Apr 14 19:44:10 CEST 2020 |
DIRECT ACCESS: http://rna.informatik.uni-freiburg.de/RetrieveResults.jsp?jobID=7212798&toolName=MutaRNA ( 30 days expiry )
Description of the job
Mutation G19U of the 5' UTR of ferritin light chain gene, which contains the iron response element (IRE); a long hairpin-stem structure. The mutation disrupts the IRE, which causes the loss of iron-level-based regulation of gene expression.
Output
download complete results
[zip]
Folding results for mutation 'G19U'
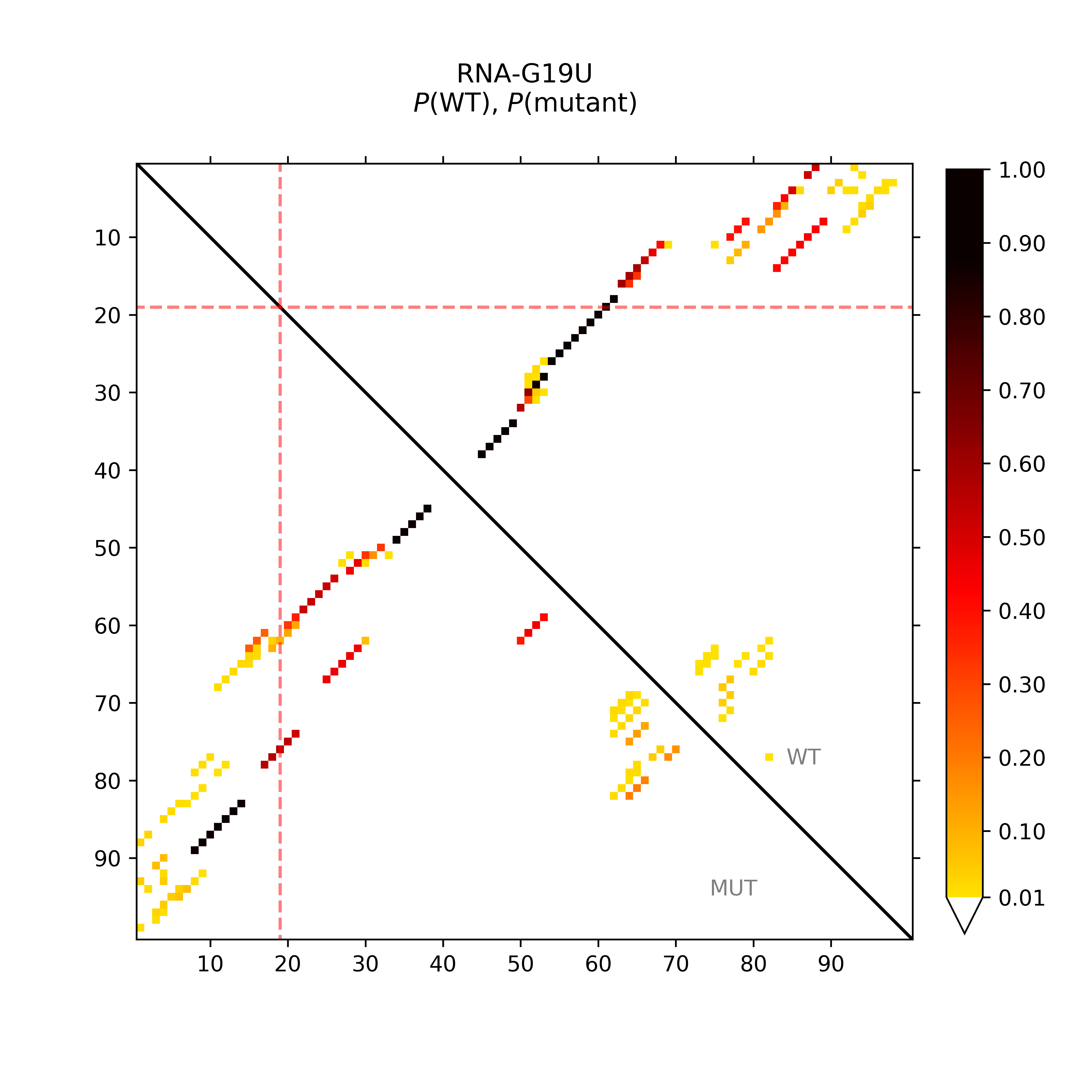
Base pairing of wildtype vs. mutant G19U
The following visualizations provide a general overview on the base pairing
potential of both the wildtype (WT) and SNP-mutated (mut) RNA sequence.
The dot plot (top-left) visualizes the base pairing potential of both the wildtype
and the mutant RNA in a heatmap-like fashion. That is the darker the dot,
the higher is the probability that the respective sequence positions form a
base pair. Probabilities derived from the Boltzmann
distributed energies of all structures that can be formed by an RNA given
the folding constraints (e.g. maximal base pair span).
The mutated position is highlighted by red dotted lines.
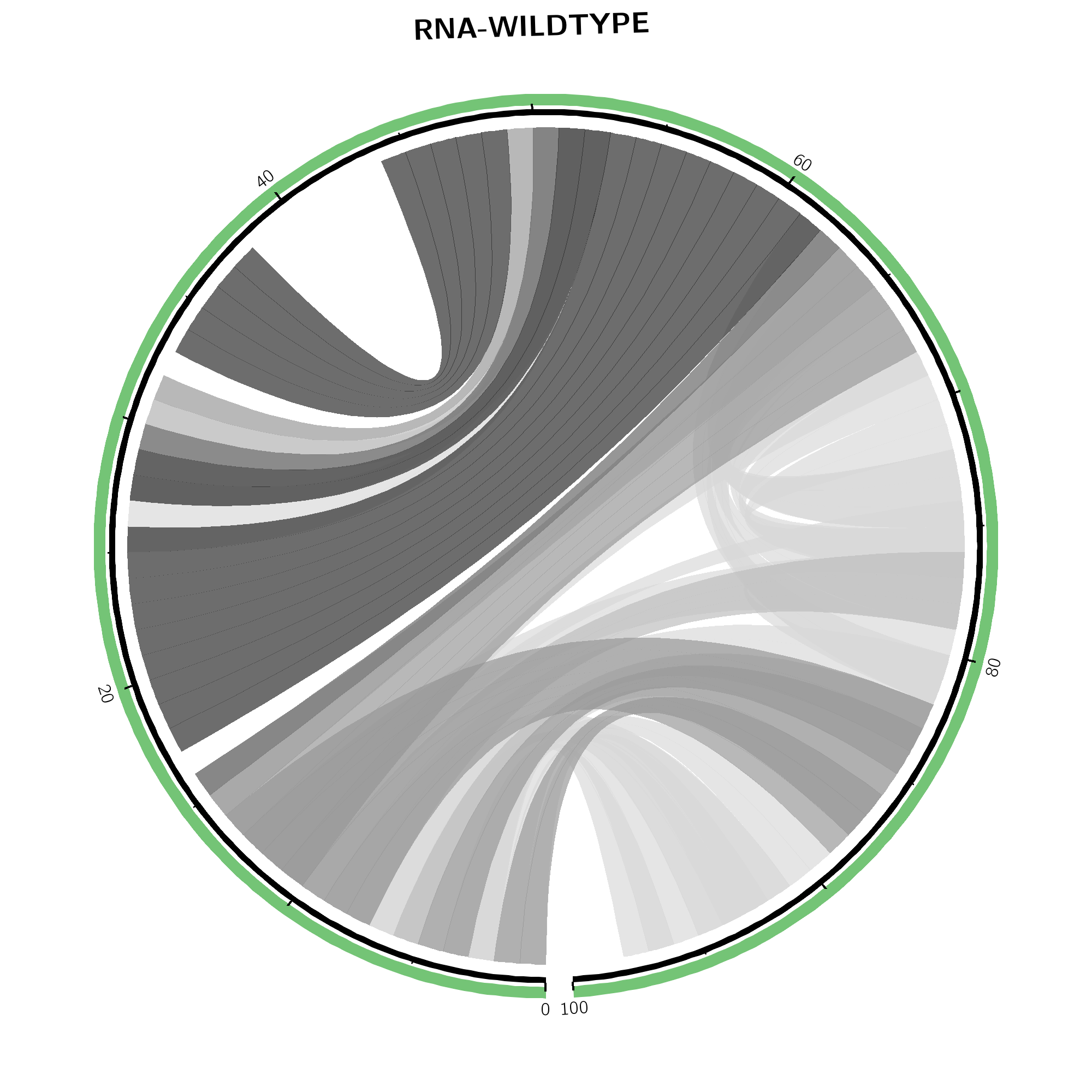
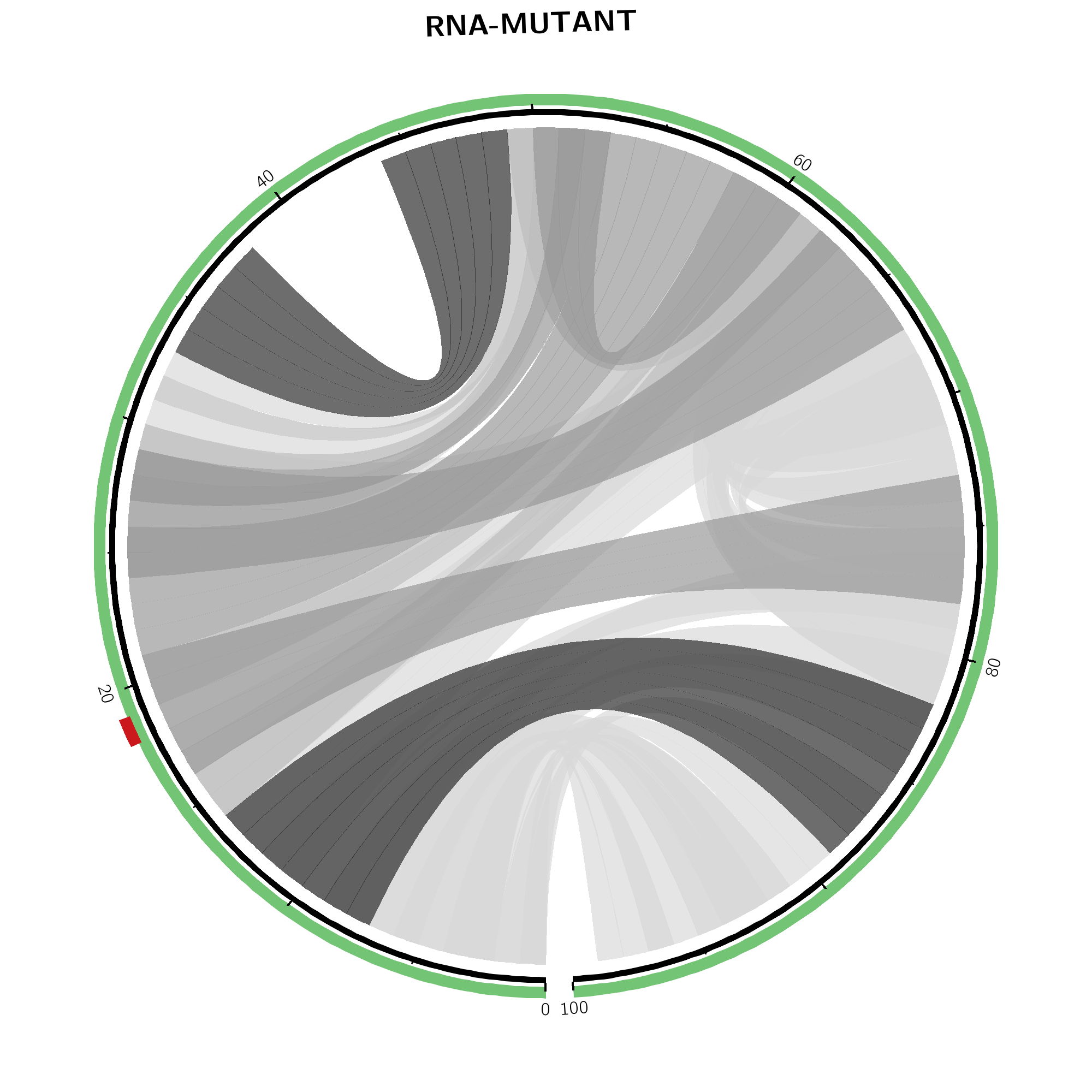
The same base pair probabilities are depicted via circular plots (top-left and -right) and arc plots (bottom-left and -right). Wildtype base pairing (top-middle) refers to the upper right part of the dot plot while mutant base pairing (top-right) covers information of the lower left part. In both, higher base pairing potential is reflected in darker hues of respective gray lines. The SNP position is marked by a red line in the outer ring within the mutant's plot on the right.
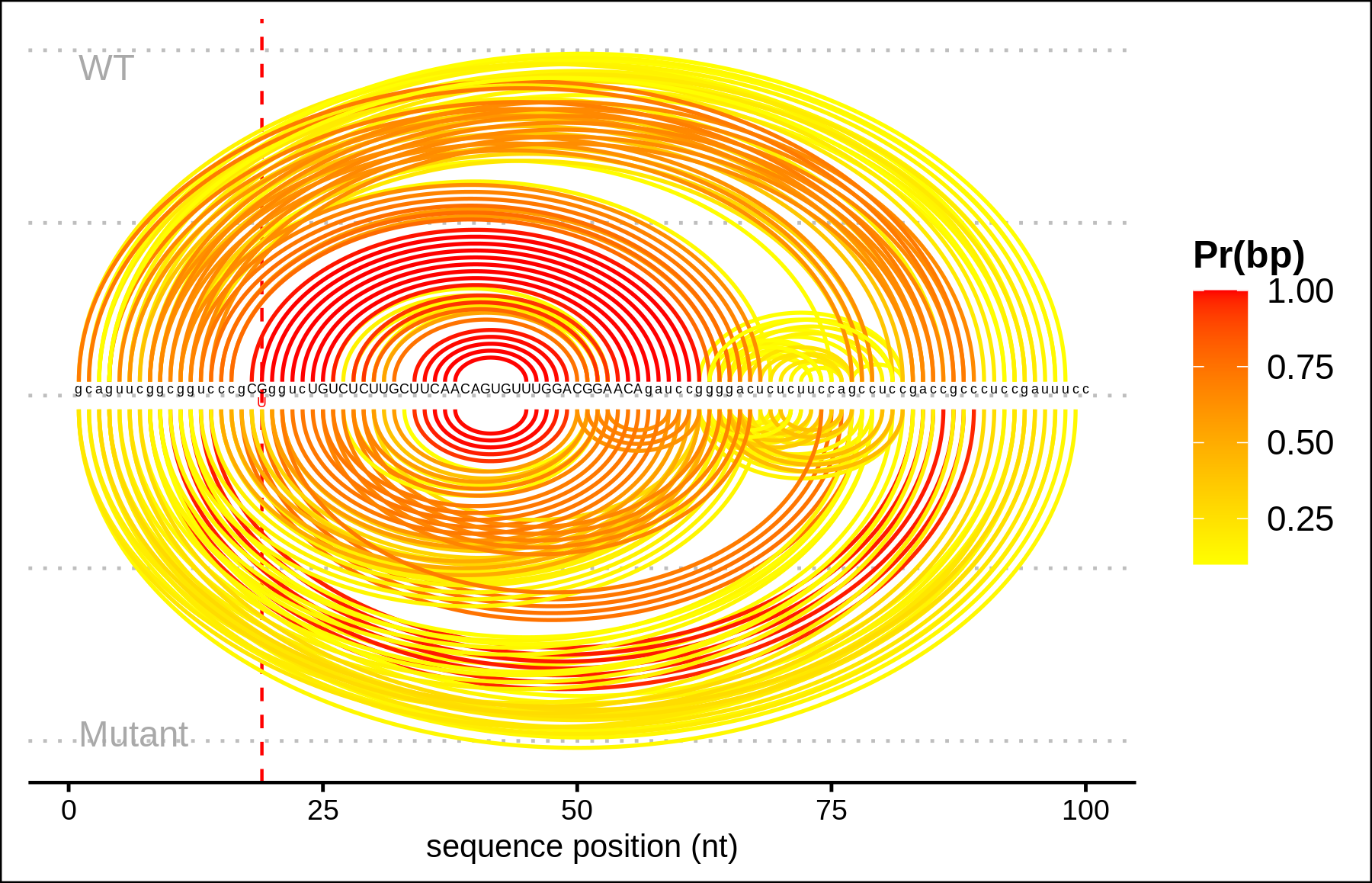
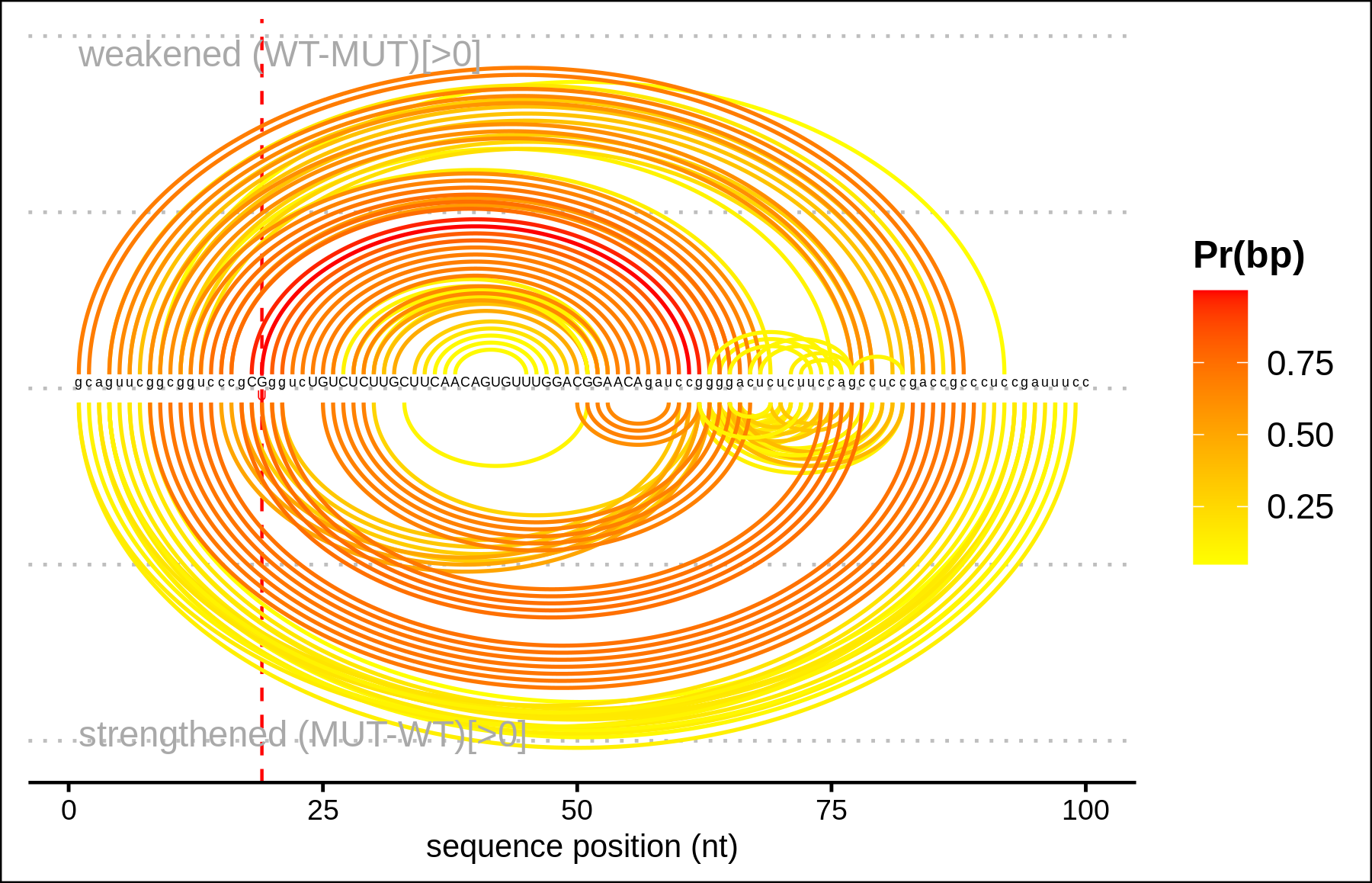
Arc plot representations (bottom) of the base pairing probabilities are analogous to the circular plots but use a heatmap-like arc coloring. That is highest probabilities are dark red while low probable base pairs are depicted in yellow.
Base pairing probabilities were extracted from the following dot plots: [WT.ps] [mut.ps] (RNAplfold (Bernhart et al., 2011)).
The same base pair probabilities are depicted via circular plots (top-left and -right) and arc plots (bottom-left and -right). Wildtype base pairing (top-middle) refers to the upper right part of the dot plot while mutant base pairing (top-right) covers information of the lower left part. In both, higher base pairing potential is reflected in darker hues of respective gray lines. The SNP position is marked by a red line in the outer ring within the mutant's plot on the right.
Arc plot representations (bottom) of the base pairing probabilities are analogous to the circular plots but use a heatmap-like arc coloring. That is highest probabilities are dark red while low probable base pairs are depicted in yellow.
Base pairing probabilities were extracted from the following dot plots: [WT.ps] [mut.ps] (RNAplfold (Bernhart et al., 2011)).
Differential comparison for G19U
To better see the often local structural changes induced by a SNP mutation,
the differences of the mutant's base pairing potential compared to the
wildtype sequence are given below. Furthermore, the effects on the
single-strandedness, i.e. accessibility, are visualized in terms of
single-position unpaired probabilities within the structural ensembles.
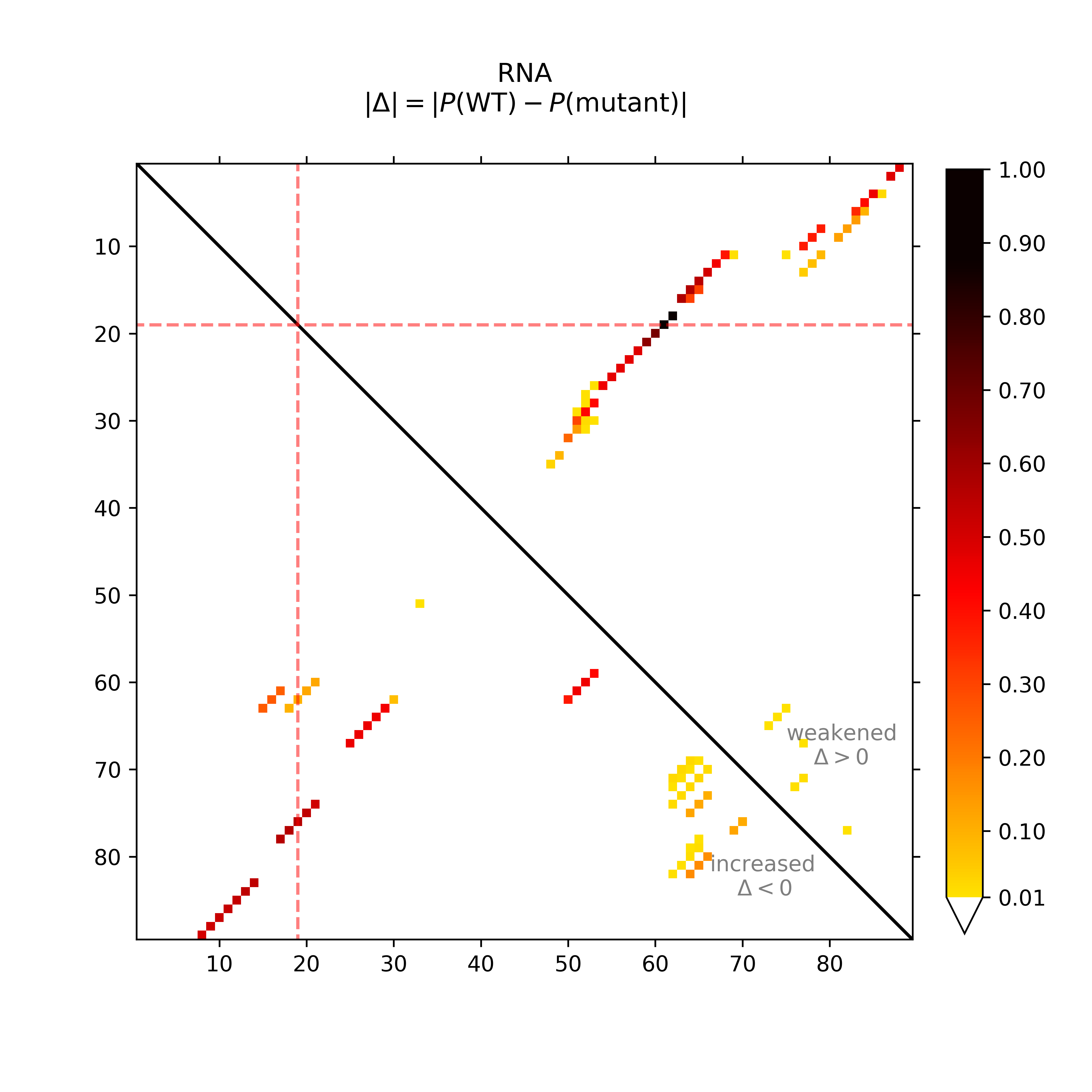
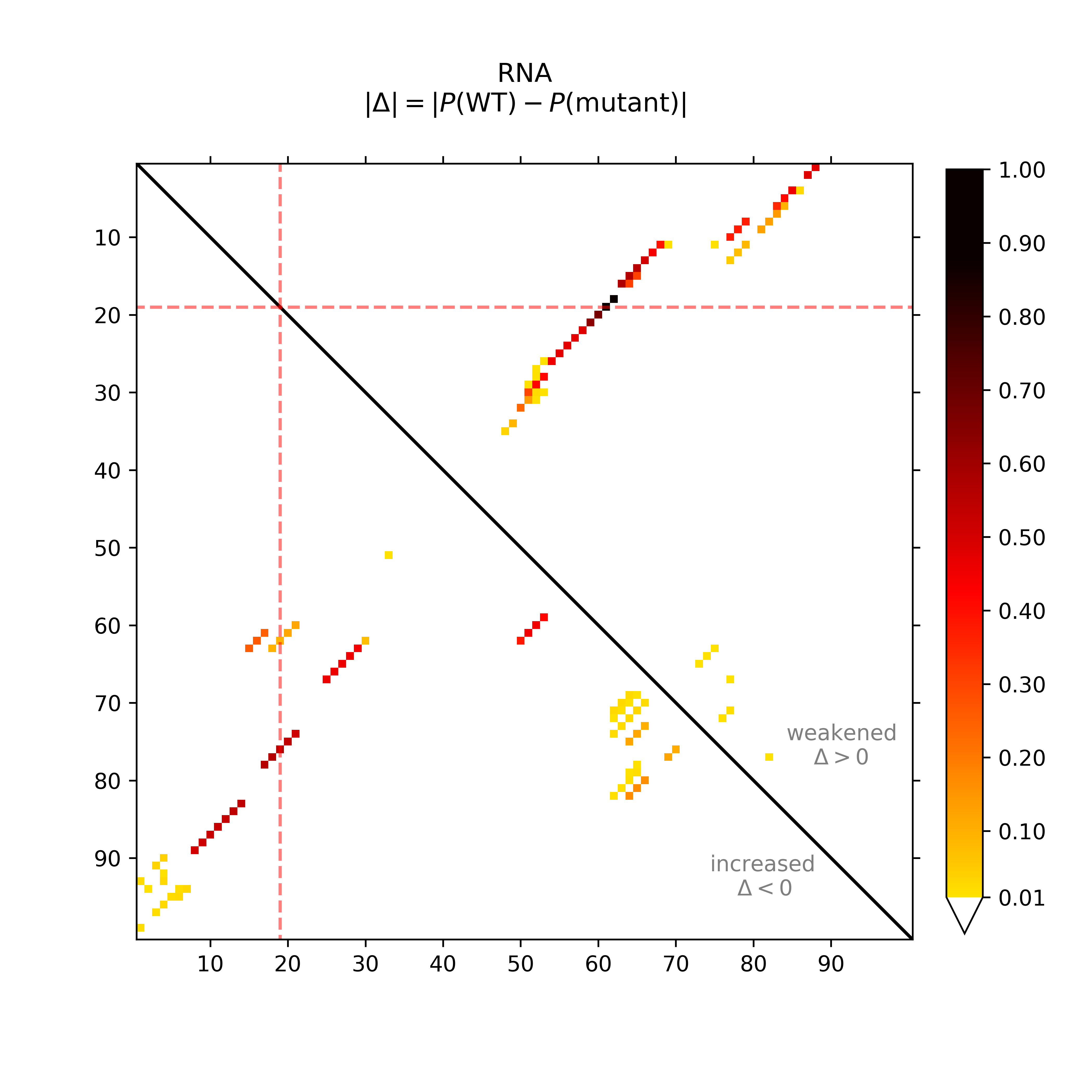
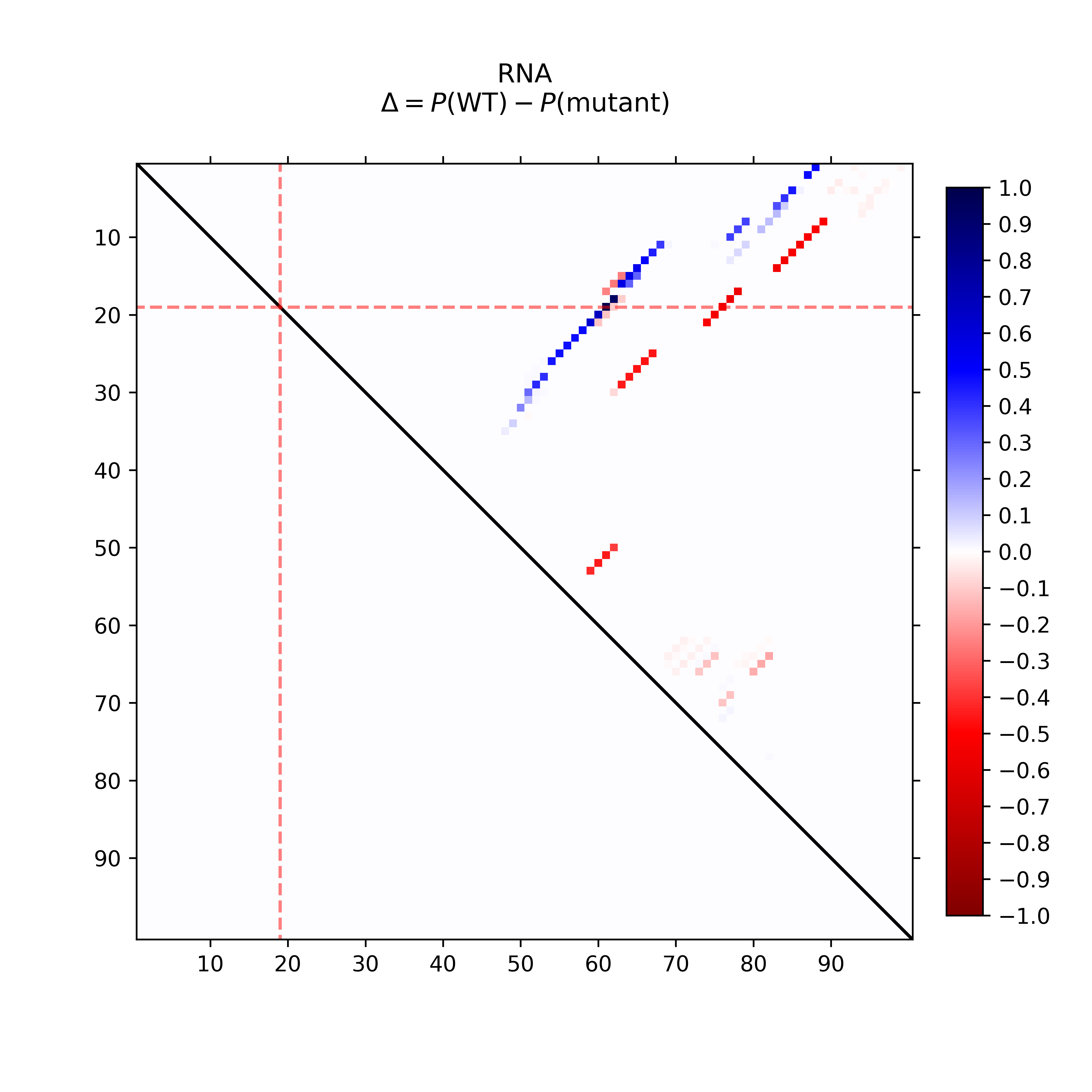
The dot plot (top-left) provides the absolute differences of the base pairing
probabilities of SNP-mutated vs. wildtype RNA, i.e. in detail Pr(bp in WT) - Pr(bp in mut).
The upper right part of (top-left) visualizes positive differences, i.e. a weakening of
the base pairing potential, while the lower left reports (absolute values)
of negative differences, i.e. base pairs with increased probability within
the mutant. Again, darker hues reflect higher absolute changes.
The mutated position is highlighted by red dotted lines.
In the (top-middle) and (top-right) respective visualizations are given in a circular plotting mode. Darker gray scales refer to higher absolute changes. The SNP position is annotated by a red bar in the outer ring.
In the (top-middle) and (top-right) respective visualizations are given in a circular plotting mode. Darker gray scales refer to higher absolute changes. The SNP position is annotated by a red bar in the outer ring.
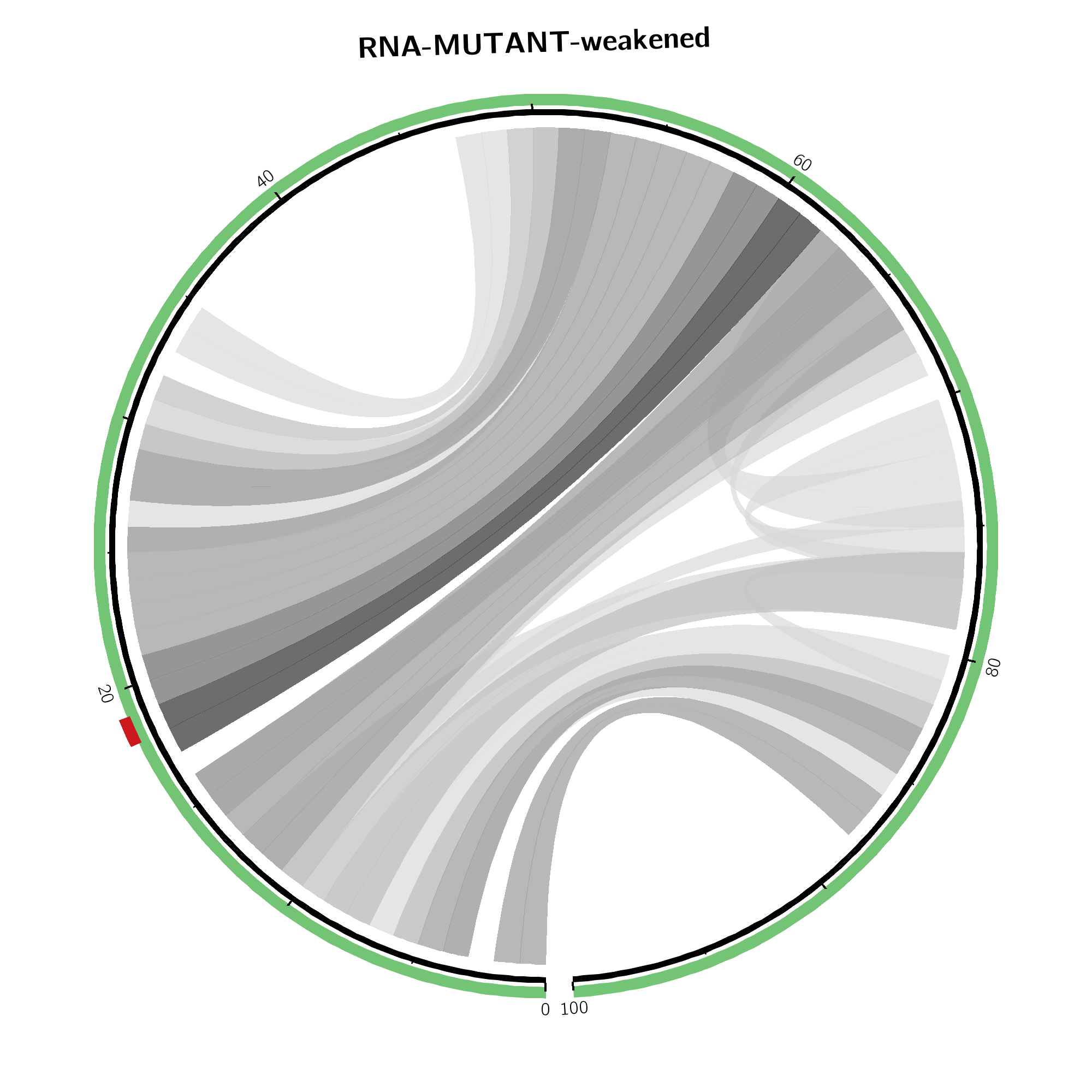
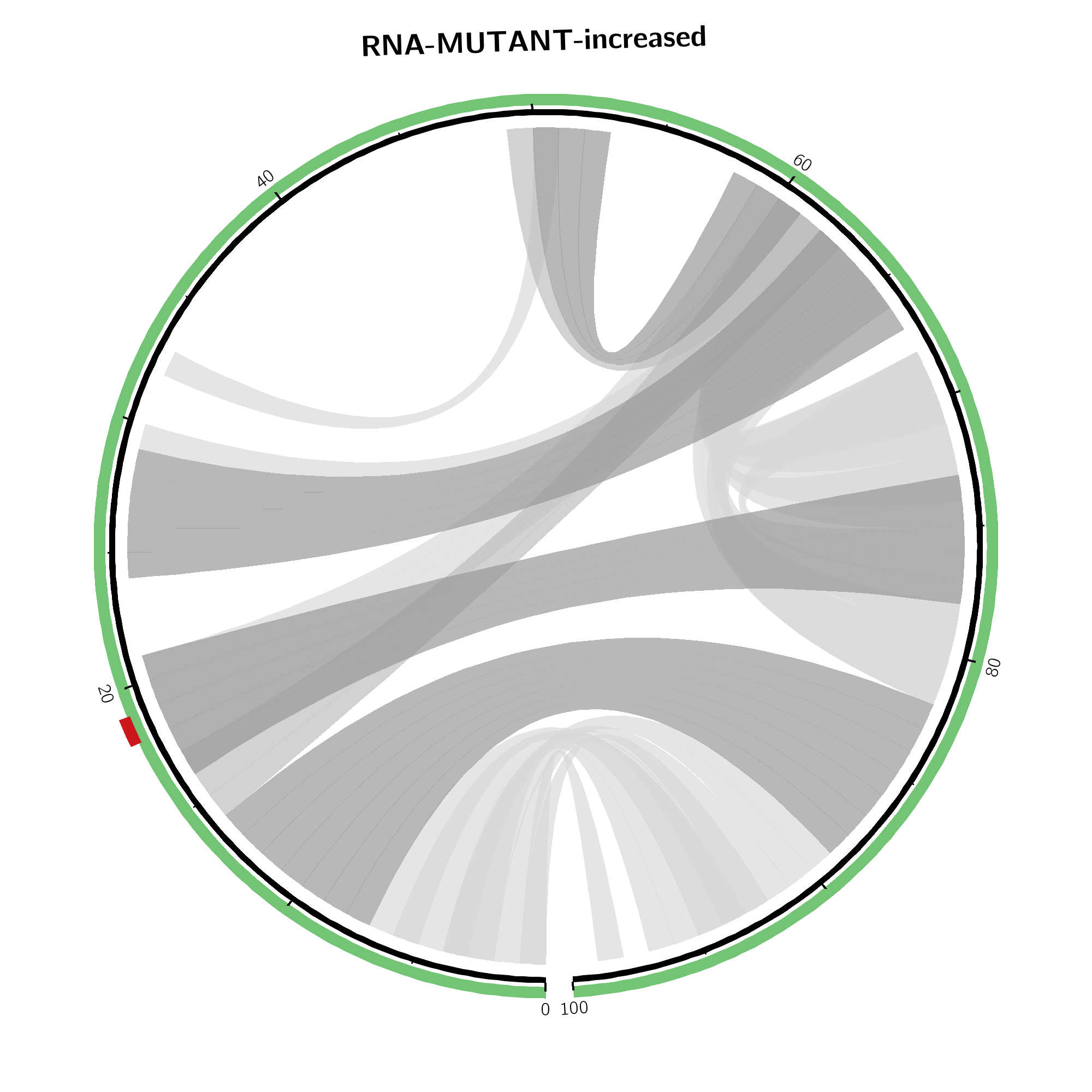
Arc plot representations (top) of the absolute change of base pairing probabilities
are analogous to the circular plots
but use a heatmap-like arc coloring. That is highest probabilities are
dark red while low probable base pairs are depicted in yellow.
Weakened base pairs are shown on top while strengthened base pairs are
depicted below the sequence. Mutations are highlighted by vertical dashed
lines.
In case the effects are local, only the respective subsequence is shown. Full length depictions are available via respective links.
In case the effects are local, only the respective subsequence is shown. Full length depictions are available via respective links.
The dot plot (left) provides the differences of the base pairing
probabilities of SNP-mutated vs. wildtype RNA, i.e. in detail Pr(bp in WT) - Pr(bp in mut).
That is base pairs weakened by the mutation are in blue while higher
base pair probability in the mutant are depicted in red.
The mutated position is highlighted by red dotted lines.
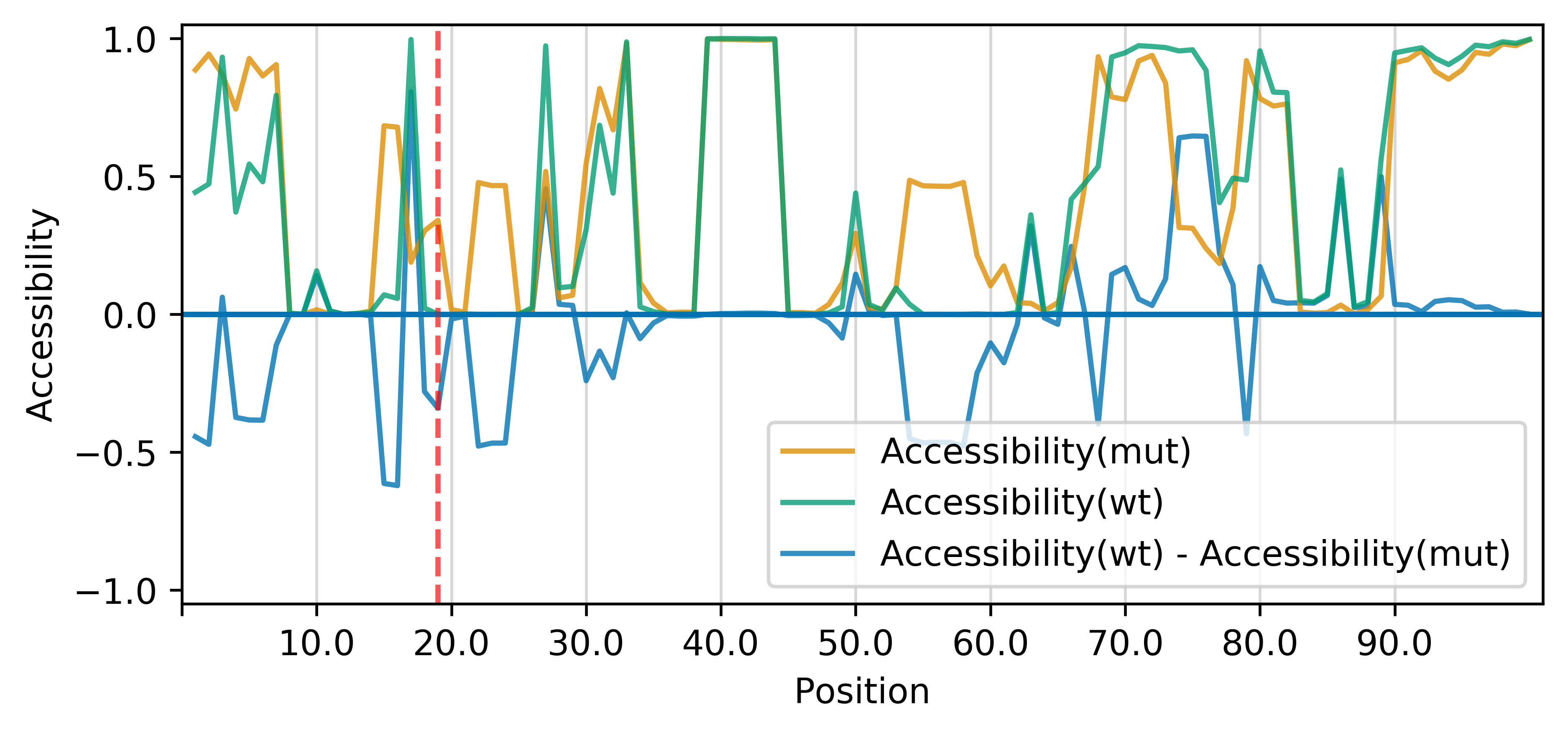
The (right) accessibility profiles of both sequences and their differences provide an assessment of the mutation's effect on the RNA's single-strandedness, which is e.g. strongly related to its interaction potential with other RNAs or proteins. Accessibility is measured in terms of local single-position unpaired probabilities (RNAplfold (Bernhart et al., 2011)); extracted from the following files: [WT.txt] [mut.txt] The mutated position is highlighted by a red line.
The (right) accessibility profiles of both sequences and their differences provide an assessment of the mutation's effect on the RNA's single-strandedness, which is e.g. strongly related to its interaction potential with other RNAs or proteins. Accessibility is measured in terms of local single-position unpaired probabilities (RNAplfold (Bernhart et al., 2011)); extracted from the following files: [WT.txt] [mut.txt] The mutated position is highlighted by a red line.
Scoring of mutation 'G19U'
Assessment using remuRNA
remuRNA (Salari et al., 2013)
computes a localized relative entropy H(WT:mut) between the Boltzmann
ensembles of the wildtype and the mutant RNAs
around the SNP of interest.
That way, effects of sequence length are reduced.
The higher remuRNA's entropy value, the stronger is the structure impact of the SNP.
If you use the score in your research, please also cite (Salari et al., 2013)
along with the version information from below.
Since remuRNA supports only SNP encodings, each SNP is scored independently when multi-nucleotide mutations are provided as MutaRNA input.
Since remuRNA supports only SNP encodings, each SNP is scored independently when multi-nucleotide mutations are provided as MutaRNA input.
Assessment using RNAsnp mode 1
RNAsnp also reported :
warnings
Assessment using RNAsnp mode 2
RNAsnp also reported :
warnings
RNAsnp (Sabarinathan et al., 2013)
focuses on the local regions of maximal structural change between mutant and wildtype RNA.
The mutation effects are quantified in terms of empirical P values.
To this end, the RNAsnp software uses extensive precomputed tables of the
distribution of SNP effects as function of length and GC content.
(Mode 1) is designed to predict the effect of SNPs on short RNA sequences (< 1000nts), where the base pair probabilities of the wildtype and mutant RNA sequences are calculated using the global folding method RNAfold. (distance) = the difference between the base pair probabilities of wildtype and mutant computed as Euclidean base pair distance. Finally, the interval with maximum base pairing distance or minimum correlation coefficient and the corresponding p-value is reported.
(Mode 2) is designed to predict the effect of SNPs on large RNA sequence using the local folding method RNAplfold with the default parameters -W 200 and -L 120. As a first step, the structural difference is calculated using the Euclidean distance measure for all sequence intervals of fixed window length. In the second step, the sequence interval with maximum base pair distance is selected to re-compute the difference for all internal local intervals. The interval with maximum base pair distance and the corresponding p-value is reported.
RNAsnp might provide additional warnings or error messages e.g. for very short sequences, which are available below the table if reported. For further details, we refer to the Help page of RNAsnp. If you use the scores in your research, please also cite (Sabarinathan et al., 2013) along with the version information from below.
(Mode 1) is designed to predict the effect of SNPs on short RNA sequences (< 1000nts), where the base pair probabilities of the wildtype and mutant RNA sequences are calculated using the global folding method RNAfold. (distance) = the difference between the base pair probabilities of wildtype and mutant computed as Euclidean base pair distance. Finally, the interval with maximum base pairing distance or minimum correlation coefficient and the corresponding p-value is reported.
(Mode 2) is designed to predict the effect of SNPs on large RNA sequence using the local folding method RNAplfold with the default parameters -W 200 and -L 120. As a first step, the structural difference is calculated using the Euclidean distance measure for all sequence intervals of fixed window length. In the second step, the sequence interval with maximum base pair distance is selected to re-compute the difference for all internal local intervals. The interval with maximum base pair distance and the corresponding p-value is reported.
RNAsnp might provide additional warnings or error messages e.g. for very short sequences, which are available below the table if reported. For further details, we refer to the Help page of RNAsnp. If you use the scores in your research, please also cite (Sabarinathan et al., 2013) along with the version information from below.
Job resubmission
 usability assessment
usability assessment

When using MutaRNA please cite :
- Milad Miladi, Martin Raden, Sven Diederichs, and Rolf Backofen
MutaRNA: analysis and visualization of mutation-induced changes in RNA structure
Nucleic Acids Research, 48(W1), W287–W291, 2020 - Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.
Results are computed with MutaRNA version 1.3.0 (using RNAplfold 2.4.14, remuRNA 1.0, RNAsnp 1.2)
About this web server
|
version 5.0.12
|
Copyright © 2012 - 2025 Bioinformatics Group Freiburg
|
|
Imprint and Disclaimer