Introduction
ExpaRNA is a fast, motif-based sequence-structure alignment
method. Instead of computing a full sequence-structure alignment,
ExpaRNA efficiently computes the best arrangement of sequence-structure
motifs common to two RNAs. A motif is a local (or isolated)
substructure which is identical to both RNAs. We call such a
substructure "exact pattern matching" (EPM).
ExpaRNA uses as a preprocessing step a previously published
algorithm from our group, which detects the set of all isolated
common substructures for two given RNAs (Backofen and Siebert,
Journal of Discrete Algorithms, 2007). Then ExpaRNA computes the
longest collinear sequence of substructures common to two RNAs.
The results of ExpaRNA are in good agreement with existing
alignment-based methods, but can be obtained in a fraction of
running time, in particular for larger RNAs.
Moreover, the ExpaRNA approach can be used to speed up
state-of-the-art Sankoff-style alignment methods like our
LocARNA algorithm and related approaches that are in principle
able to profit from alignment constraints. We have setup a
pipeline that uses the predicted sequence structure motifs as
anchor points for a LocARNA alignment. This amounts to calculate
a constraint alignment by LocARNA, which restricts the search
space and thus speeds up the alignment significantly and is
therefore important for large-scale data analysis. On the other
hand, our approach maintains existing structural motifs in the
resulting alignment.
View the
ExpaRNA tutorial video:
When using ExpaRNA please cite :
- Steffen Heyne, Sebastian Will, Michael Beckstette, and Rolf Backofen
Lightweight comparison of RNAs based on exact sequence-structure matches
Bioinformatics, 25 no. 16 pp. 2095-2102, 2009
- Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.
Results are computed with ExpaRNA version 1.0.0 linking Vienna RNA package 1.8.5
Overview
The following parameters are used to control the execution of ExpaRNA
Furthermore, additional information is available
Input Parameter
 Sequence Input in FASTA Format
Sequence Input in FASTA Format
ExpaRNA accepts input in FASTA-like format.
Input can be given either as direct text input or uploading a file.
The parameter constraints are: The input has to be in valid FASTA format. No multiline input is allowed (sequence etc. in one line each). The number of sequences has to be at least 2 and at most 2. Sequence lengths have to be in the range 1-2000. The allowed sequence alphabet is 'ACGTUNacgtun'. Fixed structure can be given in a single line with tailing '#FS' using the brace pairs (). Each sequence is allowed to have at max 0.25*seqLength 'N' characters.
Defaults to ()
Scoring Parameter
Minimal size of included substructures
Minimal size (nucleotides) of used substructures (EPMs). Minimum is 2.
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 2.
Defaults to (7)
Maximal number of used substructures
All possible substructures (EPMs) are sorted by their size, from large to small. 0 - all EPM are used 1 - the largest EPM is used 2 -...
The parameter constraints are: Input value has to be parsable as Integer. The value must be greater than or equal to 0.
Defaults to (0)
EPM scoring
1: EPM score = EPM size in Nucleotides (default)
2: EPM score = (EPM size)^2 (prefers larger patterns in LCS-EPM)
Output Parameter
Write ExpaRNA result as input for LocARNA with anchor constraints
This option create a file "LCSEPM_LocARNA_input.fa" which can be used as input LocARNA. The file contains additional anchor constraints for the LocARNA alignment
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (true)
Write ExpaRNA result as alignment into text file
This option creates a file "LCSEPM_align.aln" with the result written as alignment
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Write ExpaRNA results as list in file
This option creates a file "LCSEPM.epm" with all used substructures as list.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Write all EPMs into file
This option creates a file "allEPM.epm" with ALL common subtructures of the input RNAs.
The parameter constraints are: Input value has to be parsable as Boolean.
Defaults to (false)
Output Description
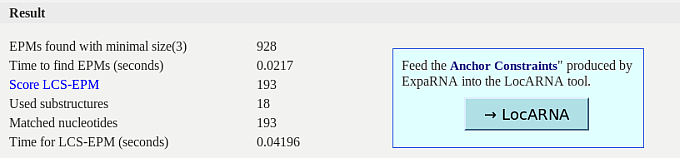
The upper part of the results shows a summary about the computed optimal set of
common substructures. The first row "EPMs found with minimal size (x)" is the overall
number of common substructures (EPMs) of size x (or larger) between the two input
RNAs. This set includes overlapping and crossing EPMs. The row "Score LCS-EPM" is
the score of the optimal set of EPMs as shown in the graphical output. The next
two rows give information about how many substructures are used in the final result
and how many nucleotides are covered by them.
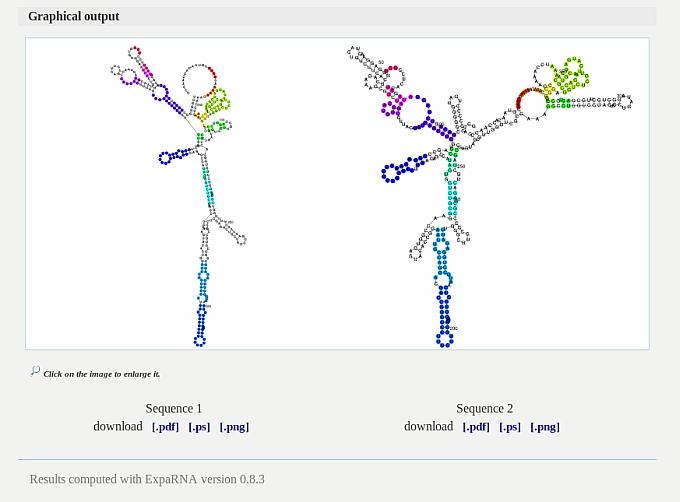
The graphical output allows to compare the input RNAs. A common substructure is
shown in the same color in both structure plots. A click on the images will
enlarge both plots for better comparison. Each annotated RNA structure can be
downloaded in different file formats (pdf,ps and png).
Input Examples
large
Alignment of two long ncRNAs with pre-defined structures.
The
example's result can be directly accessed
here small
Alignment of two relatively small ncRNAs without pre-defined structure. Here, the minimum free energy structure is computed and used for the alignment.
The
example's result can be directly accessed
hereFrequently Asked Questions
If your question is not listed, please send it to us!
 Can ExpaRNA compare multiple RNAs?
Can ExpaRNA compare multiple RNAs?
Currently, only pairwise comparison is supported. Maybe in future a multiple comparison is possible.
What is the difference between the scoring 'default' and 'prefer larger substructures'?
With the default scheme (t=1) ExpaRNA tries to find a set of common substructures with maximal number of nucleotides. With the alternative scheme (t=2), the score of each substructure is quadratic to its size in nucleotides. For example, a base pair (2 nucleoides) has score 4, two base pairs have score 16 and so on. This prefers larger substructures in the result.
Can ExpaRNA find substructures with mismatches?
No, ExpaRNA only finds exact matching substructures. Both sequence and structure of all nucleotides have to be exactly the same. If you are interested in a pure structural comparison you can change all nucleotides to 'N' and use the structure of the original sequence.
Can ExpaRNA do a structural comparison only?
Yes, to some extend. As a trick you can change all nucleotides to 'N' in combination with the structure of the original sequence.
List of Changes
- 3.0.5 : FASTA headers are not allowed to show ";/\*"
