
Input and runtime details for job 3326122 (precomputed example)
Input Parameter
 Sequence Input in FASTA Format Sequence Input in FASTA Format | [.fa] | ||
| Upload dot plots | not provided | ||
| Predict dot plots | with pseudoknots (NUPACK) |
Scoring Parameters
| Structure Weight | 200 | ||
| Indel Opening Score | -500 | ||
| Indel Score | -350 | ||
| Use RIBOSUM | yes | ||
| Match Score | 50 | ||
| Mismatch Score | 0 |
Heuristics for speed/accuracy tradeoff
| Minimal Pair Probability | 0.01 | ||
| Maximal Difference for Sizes of Matched Arcs | 30 | ||
| Maximal Difference for Alignment Edges | 60 |
Other Parameter
| Ignore Constraints | no | ||
| Search Time Limit (in milliseconds) | 300000 | ||
| Disallow Lonely Pairs | yes |
Job ID 3326122 (server version trunk)
 | Job Submitted & Queued | @ Fri Jul 22 11:59:54 CEST 2016 | |
| CARNA Started | @ Fri Jul 22 11:59:56 CEST 2016 | |
| CARNA Finished & Post-Processing | @ Fri Jul 22 12:00:01 CEST 2016 | |
| Post-Processing Finished | @ Fri Jul 22 12:02:00 CEST 2016 | |
| Job Completed | @ Fri Jul 22 12:02:15 CEST 2016 |
Description of the job
pseudoknots
This example demonstrates CARNA's capability to align RNA with pseudoknots. In this example, we provide fixed input structures with pseudoknots. Thereby, we demonstrate the syntax of constraint annotation in the fasta file. In the output of this example correct alignment of the pseudoknots is best seen from our conservation consensus dot plot representation. Please note that the consensus structure in the shown alignment (Alignment annotated with pseudoknot-free consensus structure) does not show the pseudoknot because this consensus structure is generated by RNAalifold from the CARNA alignment. RNAalifold was not designed to predict pseudoknots. Since we provide fixed structures in this example, it runs with default settings. To predict pseudoknots from ensembles, one has to explicitly predict the ensemble dot plots with pseudoknots. This is supported via a tool from NUPACK. Due to the hardness of pseudoknot folding, this will work for only comparably simple pseudoknots as described by Dirks and Pierce (J Comput Chem, 2004).
Outputdownload complete results [zip]
Conservation Dot Plots
Loading image 

 Click on the dot plot to enlarge it.
Click on the dot plot to enlarge it.
![[png]](/DownloadFile.jsp?jobID=3326122&toolName=CARNA&fileNameOrType=input.out/plots/average_dlin_025.png){kind=link}
![[png]](/DownloadFile.jsp?jobID=3326122&toolName=CARNA&fileNameOrType=input.out/plots/average_dlin_05.png){kind=link}
For interpretation of the colors see the legend below. For a detailed description of the output, please see the help page.
Color Legend
The lower left triangle of the dot plots contains the average dot plot colored with variance information. Pure green means maximum variance (e.g. in half of the sequences the dot has probability 0 and in the other half it has probability 1); pure red means no variance at all (the dot has the same probability in all sequences).

Alignment annotated with pseudoknot-free consensus structure
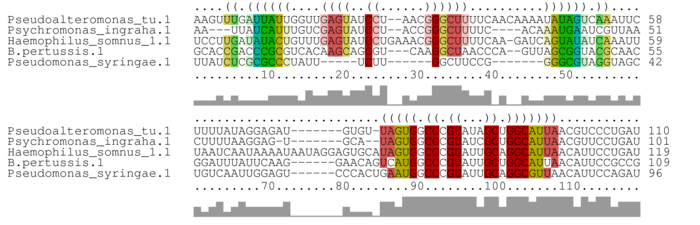
Bases are encoded by IUPAC codes. For interpretation of the colors see the legend below. For a detailed description of the output, please see the help page.
Color Legend
Job resubmission
 usability assessment
usability assessment

When using CARNA please cite :
- Dragos A. Sorescu, Mathias Moehl, Martin Mann, Rolf Backofen, and Sebastian Will
CARNA - alignment of RNA structure ensembles
Nucleic Acids Reseach, 2012, 40 no. W1 pp. W49-W53 - Alessandro Dal Palu, Mathias Moehl, Sebastian Will
A Propagator for Maximum Weight String Alignment with Arbitrary Pairwise Dependencies
Proceedings of the 16th International Conference on Principles and Practice of Constraint Programming (CP-2010), 2010, 8 - Martin Raden, Syed M Ali, Omer S Alkhnbashi, Anke Busch, Fabrizio Costa, Jason A Davis, Florian Eggenhofer, Rick Gelhausen, Jens Georg, Steffen Heyne, Michael Hiller, Kousik Kundu, Robert Kleinkauf, Steffen C Lott, Mostafa M Mohamed, Alexander Mattheis, Milad Miladi, Andreas S Richter, Sebastian Will, Joachim Wolff, Patrick R Wright, and Rolf Backofen
Freiburg RNA tools: a central online resource for RNA-focused research and teaching
Nucleic Acids Research, 46(W1), W25-W29, 2018.